










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Biológica Colombiana
Print version ISSN 0120-548X
Acta biol.Colomb. vol.21 no.1 Bogotá Jan./Apr. 2016
https://doi.org/10.15446/abc.v21n1.49761
Doi: http://dx.doi.org/10.15446/abc.v21n1.49761.
THE HUMAN MICROBIOTA: THE ROLE OF MICROBIAL COMMUNITIES IN HEALTH AND DISEASE
La microbiota humana: comunidades microbianas en la salud y en la enfermedad
Luz Elena BOTERO1,2, Luisa DELGADO-SERRANO3,4, Martha Lucía CEPEDA HERNÁNDEZ3, Patricia DEL PORTILLO OBANDO3, María Mercedes ZAMBRANO EDER3.
1 Facultad de Medicina, Universidad Pontificia Bolivariana. Calle 78B no. 72A-109, Bloque B, Piso 5. Medellín, Colombia.
2 Unidad de Bacteriología y Micobacterias, Corporación para Investigaciones Biológicas, Unidad Pontificia Bolivariana. Carrera 72A no. 78B-141. Medellín, Colombia.
3 Corporación Corpogen. Carrera 5 no. 66A-34. Bogotá D.C., Colombia.
4 Centro de Bioinformática y Biología Computacional- BIOS. Carrera 15B no. 161. Manizales, Colombia.
For correspondence. luz.boterop@upb.edu.co
Received: 22nd March 2015, Returned for revision: 14th April 2015, Accepted: 10th July 2015.
Associate Editor: Nubia Estela Matta Camacho.
Citation / Citar este artículo como: Botero LE, Delgado-Serrano L, Cepeda ML, Del Portillo P, Zambrano MM. The human microbiota: the role of microbial communities in health and disease.Acta biol. Colomb. 2016;21(1):5-15. doi: http://dx.doi.org/10.15446/abc.v21n1.49761.
ABSTRACT
During the last decade, there has been increasing awareness of the massive number of microorganisms, collectively known as the human microbiota, that are associated with humans. This microbiota outnumbers the host cells by approximately a factor of ten and contains a large repertoire of microbial genome-encoded metabolic processes. The diverse human microbiota and its associated metabolic potential can provide the host with novel functions that can influence host health and disease status in ways that still need to be analyzed. The microbiota varies with age, with features that depend on the body site, host lifestyle and health status. The challenge is therefore to identify and characterize these microbial communities and use this information to learn how they function and how they can influence the host in terms of health and well-being. Here we provide an overview of some of the recent studies involving the human microbiota and about how these communities might affect host health and disease. A special emphasis is given to studies related to tuberculosis, a disease that claims over one million lives each year worldwide and still represents a challenge for control in many countries, including Colombia.
Keywords: 16S rRNA gene, microbial diversity, microbiome, Mycobacterium tuberculosis.
RESUMEN
En las últimas décadas ha incrementado nuestro conocimiento sobre la gran cantidad de microorganismos que conviven con nosotros, comunidades que colectivamente se conocen como la microbiota humana. El número de microorganismos que conforman la microbiota supera el número de células del cuerpo humano por un factor de diez aproximadamente y aporta un gran repertorio de genes y procesos metabólicos. La diversidad de la microbiota humana y su potencial metabólico brindan al hospedero una serie de funciones que complementan sus procesos y a su vez pueden influir sobre la salud del ser humano en formas que apenas se empiezan a conocer. La microbiota varía desde el nacimiento hasta la vejez del individuo, con características que dependen del sitio corporal, del estilo de vida y del estado de salud del hospedero. El reto actual es aprovechar el conocimiento derivado de la identificación y caracterización de estas comunidades microbianas para entender cómo funcionan estos microorganismos y cómo pueden influir de forma positiva o negativa sobre la salud del humano. En este documento ofrecemos una revisión general de algunos estudios recientes sobre la microbiota humana y su posible efecto en el hospedero en términos de salud y bienestar. Igualmente, se mencionan estudios sobre microbiota y su posible asociación con la tuberculosis, una enfermedad que todavía cobra más de un millón de vidas anualmente a nivel mundial y cuyo control todavía representa un gran reto en varios países del mundo, incluido Colombia.
Palabras clave: 16S ARNr, diversidad microbiana, microbioma, Mycobacterium tuberculosis.
INTRODUCTION
Microorganisms are known for their capacity to inhabit and thrive in a broad range of natural environments. Over the last decade, research has shown that diverse and abundant microbes are associated with humans, forming microbial communities called the human microbiota. The human microbiota is also quite diverse and inhabits different locations in the body, some external such as the skin, and others internal like the mucosal epithelia of the intestine, vagina, or the respiratory tract. The microbiota, which consists of viruses and unicellular microbes, mainly Bacteria and Archaea, as well as eukaryotes (fungi and protozoa) (Clemente et al., 2012; Belkaid and Hand, 2014), has been linked to a wide range of phenotypes associated with health and disease. Technological and molecular developments, such as PCR amplification and sequencing, from Sanger sequencing methods to novel high-throughput massive parallel techniques, have revealed not only that the human microbiota varies in composition, but can also carry out metabolic functions that are absent in human host cells (Gill et al., 2006). Thus, in recent years we have started to fully appreciate and understand the importance of these metabolically diverse communities within the host.
The human host is estimated to harbor up to 1014 microbes, mainly bacteria in the intestine, which outnumber human cells by a ratio of ten to one (Ursell et al., 2012; Belkaid and Hand; 2014). In turn, the human microbiome, which is the entire genomic content of the host-associated microbiota, contains a genetic repertoire that is larger than that of the human genome and thus can considerably extend the host's metabolic capacity. Due to this large number of microbial cells, the human can be considered as a "meta-organism" (Ley et al., 2008; Belkaid and Hand, 2014) consisting of its own cells and microorganisms that co-exist in a relationship that, in most cases, can be mutually beneficial, but in others can result in disease (Turnbaugh et al., 2007; Clemente et al., 2012). Together, the human and its microbiota form a complex ecosystem involving multiple biological activities that result in a homeostatic relationship important for human health and disease (Costello et al., 2012). This diverse group of microbial organisms, their genes and their associated metabolic processes impact host health by playing important roles in key aspects such as the maturation of the immune system, defense against pathogens, access to complex nutrients and degradation of toxic compounds. The complexity of these communities and their interaction with the host is illustrated by the fact that both beneficial and pathogenic microorganisms can be part of our microbiota, and that in some cases altering microbial community members can lead to the development of inflammatory processes that have negative consequences for the host (Belkaid and Hand, 2014; Glendinning and Free, 2014; Silva et al., 2015).
From the moment of birth, each individual is colonized by a variety of microorganisms that constitute a vital part of the host throughout his lifetime. The selection and establishment of a stable microbiota in individuals with different lifestyles and living conditions is currently of considerable research interest. There is emerging evidence that the microbiota modulates a number of processes that result in the development, maturation, differentiation and proliferation of the mucous membranes, both at the cellular and molecular levels (Sharma et al., 2010). The microbiota can set off a chain of molecular events that play a key role in the maturation of the innate and adaptive immune systems and thus, has a profound effect on mucosal barriers such as that of the intestinal mucosa (Sharma et al., 2010). Disruption of the human-microbiota homeostatic interactions are associated with clinical disorders such as inflammatory diseases of the gastrointestinal tract, asthma, periodontitis, vaginal and antibiotic-induced Clostridium difficile infections, as well as obesity and malnutrition (Costello et al., 2012; Silva et al., 2015). The microbiota is also known to provide protection against potential pathogens. This protection is achieved via mechanisms such as competition for nutrients, production of antimicrobial compounds or induction of these antimicrobials by host cells, promotion of changes that make host environments hostile for pathogen establishment, and modulation of the immune system that promotes responses against pathogens (Belkaid and Hand, 2014). Understanding the processes that lead to the assembly and disruption of the complex host-microbiota relationship are fundamental for future efforts aimed at maintaining a symbiotic balance key for health and well-being. Viewing the host in the context of its interactions with the resident microbial communities, and not just as a host that battles against disease, can encourage management of health and treatment of certain diseases via restoration of host-microbiota equilibrium (Costello et al., 2012; Lemon et al., 2012; Dore and Blottiere, 2015).
TECHNIQUES FOR ANALYSIS OF MICROBIAL COMMUNITIES
The microbiota was traditionally studied by staining techniques of samples obtained from different body surfaces. These include the Gram stain, which has been extremely valuable for distinguishing many bacterial groups but lacks taxonomic resolution. The use of culture techniques then allowed the elucidation of phenotypic characteristics of isolated microorganisms, including nutritional requirements and production or consumption of metabolic compounds. Thus, growing organisms in the laboratory has been essential for microbial identification and characterization. These analyses, however, are restricted to those organisms that can grow under in vitro conditions, which represent only a minority of microbial species, a shortcoming that has been overcome with the use of culture-independent methods based on DNA (Morgan and Huttenhower, 2012). More recently, the use of molecular approaches and massive sequencing techniques that provide fast analyses and lower costs have led to significant progress in the study of human-associated microbial communities.
Culture-independent techniques do not rely on obtaining cultured microorganisms but rather use the total DNA extracted directly from a sample to probe microbial communities. The analysis of this metagenomic DNA can uncover microorganisms that have not yet been isolated in the laboratory, possibly due to failure in meeting the conditions required for in vitro growth. Two approaches are currently used for characterization of human-associated microbial communities. The first one is based on amplification of conserved genes such as the 16S rRNA gene that codes for the small ribosomal subunit in Bacteria and Archaea. Analysis of these genes permits phylogenetic identification based on comparison with sequence databases to quickly determine the lineages present in a sample (Hamady and Knight, 2009). Another approach involves isolation, sequencing and analysis of the entire DNA present in a particular environment, known as the metagenome. While the first strategy can reveal the microbial community composition and structure based on taxonomy, metagenomic studies provide information not only of the members of a microbial community but also about the metabolic functions encoded in these microbial genomes (Handelsman, 2004; Wang et al., 2015).
Modern massive parallel sequencing platforms (such as 454, Illumina and Ion Torrent) are based on the use of barcodes to distinguish samples which are sequenced simultaneously, providing great volumes of data of millions of reads, and a much greater coverage in comparison to traditional Sanger sequencing. For taxonomic profiling of communities, 16S rDNA regions are typically PCR-amplified, labeled with barcodes and sequenced; the sequences generated are then processed using bioinformatic workflows for identification and analysis. Some of the most common bioinformatic tools used for microbial community analysis include the Ribosomal Database Project-RDP (Cole et al., 2009; Cole et al., 2014), the Mothur software (Schloss et al., 2009), and the Workflow for Alignment, Taxonomy and Ecology of Ribosomal Sequences-WATERS (Hartman et al., 2010) and the bioinformatics pipeline for Quantitative Insights Into Microbial Ecology QIIME (Caporaso et al., 2010). After filtering reads for quality and separating samples by barcode, sequences are usually grouped into operational taxonomic units (OTUs) based on the level of sequence identity, which for 16S rDNA is usually 97 % or higher, although this cutoff is still under debate (Gevers et al., 2005; Kim et al., 2014). OTUs are then compared to freely available curated 16S rDNA sequences in databases such as Greengenes (DeSantis et al., 2006) or RDP (Cole et al., 2009), in order to obtain taxonomic identification. In addition, the ability to compute data in the "cloud", like the Amazon Web Services (AWS) Elastic Compute Cloud (EC2), means that any researcher with access to internet can be connected to a supercomputer and can analyze hundreds of millions of sequences at a relatively low cost (Ursell et al., 2012).
THE MICROBIOTA AND ITS EFFECT ON THE HUMAN HOST
The interest in characterizing the human microbiota stems from a desire to understand how it may be associated with a specific condition or may influence susceptibility to or development of a disease (Zaneveld et al., 2008). Table 1 lists some of the body sites that have been characterized and studied regarding their possible association with health and/or disease. The growing importance of this field is reflected in major initiatives such as the Human Microbiome Project of the National Institute of Health in the United States (http://commonfund.nih.gov/index), the Metagenomics of the Human Intestinal Tract project-MetaHIT funded by the European Commission (2008-2012) ( http://www.metahit.eu/) and the International Human Microbiome Consortium (2011-2015) (http://www.human-microbiome.org/index.php). These international efforts intend to expand our knowledge regarding these communities with the aim of understanding various aspects about the interactions of microorganisms with human biology. It is expected that in the longer term the knowledge gained from these studies can generate useful information for addressing health issues, such identification of specific markers, signaling molecules or enzymes associated with or that can contribute to human well-being (Turnbaugh et al., 2007; Group et al., 2009; Proctor, 2011).
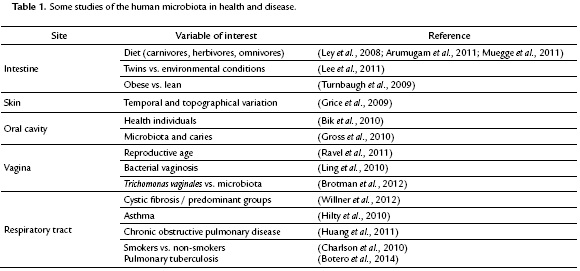
Despite the great number of studies conducted so far, it remains unclear whether observed changes in the microbiota are a consequence of a particular health/disease condition or contribute to its development. To address this question, it is necessary to establish the microbiota of healthy individuals in order to first identify variations associated with temporal changes, ethnic or cultural differences, among other factors, which can then allow us to accurately assess whether observed changes are actually associated with a diseased state (Ursell et al., 2012). We briefly describe below some findings that demonstrate the interesting developments regarding the microbiota in different body sites, including the intestine, vagina, skin, oral cavity and respiratory tract.
Intestinal microbiota
Early research on the human microbiota focused on the study of intestinal communities, probably the largest and most complex human-associated community. These studies revealed for the first time the extent of the collective microbial repertoire, with microbial cells outnumbering human cells by at least a factor of 10 (Ursell et al., 2012; Belkaid and Hand, 2014). Microbial cells were also found to enrich functional capabilities absent in the human genome. They can produce nutrients and metabolites and enable the use of nutrients that otherwise would not be digestible and could even adversely affect host health status (Gill et al., 2006; Wu et al., 2011; Clemente et al., 2012; Dore and Blottiere, 2015). Given the large microbial diversity described in environmental ecosystems (at least 55 divisions belonging to Bacteria and 13 to Archaea) and the high species-level difference found among individuals, it is interesting to find an overall predominance of a few groups at the gut level: two bacterial phyla (Firmicutes and Bacteroidetes) and one archaeon (Methanobrevibacter smithii). The microbial composition also varies along the gastrointestinal tract.
Samples taken of the mouth, stomach, duodenum, colon and stool of four healthy individuals (two men and two women) revealed that the mouth harbored the greatest phylogenetic diversity whereas the stomach was the least diverse (Stearns et al., 2011). Gut communities, with their large metabolic capacity, can also have unexpected consequences on the host. Changes in diet, for example, alter the intestinal microbiota and have recently been correlated with shifts in patterns associated with cancer risk, such as inflammation of the mucosa and production of metabolites in a study involving rural Africans and African-Americans (O'Keefe et al., 2015). Given the broad range of interactions with the human, from providing additional metabolic functions to modulation of the immune system and even effects on host behavior, the intestinal microbiota has even been considered as an additional organ within the human host (Glendinning and Free, 2014).
The fact that particular microbial groups are maintained among humans most probably reflects selection and a process of co-evolution between the host and its microbial communities (Ley et al., 2006). In this regard, the host diet is considered the major factor influencing the microbial communities in healthy individuals. A study of gut microbial communities in humans and other mammals indicated an existing core microbial community based on diet (carnivores, herbivores and omnivores), in most cases independent of host phylogeny (Ley et al., 2008). Other studies have focused on obesity, finding that it can be linked to changes in the community at the phylum level, reduction in bacterial diversity and alteration of particular genes and metabolic pathways (Turnbaugh et al., 2009). Recent work has shown that alterations of the gut microbiota caused by consumption of artificial sweeteners such as saccharine can lead to glucose intolerance (Suez et al., 2014). In addition, the use of antibiotics, specifically during development in early infancy has been postulated to result in disruption of the microbiota, which in turn can result in alterations of host metabolic functions with long-lasting consequences for health, such as weight increase and obesity (Cox and Blaser, 2015).
Microbiota of the vagina
The vaginal microbial communities have long been considered an important defense mechanism against infections (Ma et al., 2012). Studies of women of different ethnic backgrounds, including Caucasians, African Americans, Hispanics and Asians, showed that most communities are dominated by a single Lactobacillus species in each group of women, the most common being L. iners, L. crispatus, L. gasseri and L. jensenii (Zhou et al., 2007; Ravel et al., 2011). These results indicate possible metabolic differences that may alter the levels of lactic acid production and thus provide levels of protection that can vary in each ethnic group. In contrast to healthy women, there is an increase in the richness and diversity of genera and species in women with bacterial vaginosis. Longitudinal studies have shown that the vaginal microbiota can be highly dynamic, with drastic changes in bacterial composition and abundances in response to factors such as hormones and antibiotics. The recurrence of bacterial vaginosis has been associated with either reinfection or reemergence of endogenous microorganisms, a situation that is favored by hormone fluctuations, antibiotic treatments and intrauterine devices, among others (Fredricks, 2011). The vaginal microbiota has also been found to be important for colonization and development of the neonatal microbiome, which is determined in large part by the maternal-offspring exchange during delivery. Disruption of this exchange, as occurs with Cesarian sections and use of antibiotics, can lead to increased risk of various diseases such as obesity and diabetes (Mueller et al., 2015).
Oral microbiota
The microbiota of the oral mucosa in healthy individuals shows variability among individuals, probably due to changing environmental factors, nutrition, hygiene and genetic conditions that favor establishment and predominance of particular microorganisms in each individual (Bik et al., 2010). The oral microbiota is a complex community composed of many species, some of which can form biofilms and lead to gum disease, as is the case of the intracellular opportunistic pathogen Porphyromonas gingivalis. Conversely, other inhabitants of the oropharynx, such as Streptococcus viridans, can produce antibacterial compounds that prevent colonization of pathogenic microorganisms such as Streptococcus pneumoniae and Streptococcus pyogenes. However, the use of broad-spectrum antibiotics can indiscriminately eliminate resident communities, including beneficial microorganisms, and allow growth of pathogenic microorganisms. Given the importance of the oral microbiota to host health status, new efforts have focused on describing and understanding the dynamics of these populations at greater depth (Bik et al., 2010; Chen and Jiang, 2014). A study of ten healthy individuals showed that 15 genera were common to all samples. However high variability was also detected among samples with predominance of the genera Streptococcus, Prevotella, Neisseria, Haemophilus and Veillonella (Bik et al., 2010). Another study carried out by Huang (2011), was able to distinguish between microbial communities of the dental plaque of healthy individuals and those with chronic gingivitis, identifying eight taxa associated to the disease (Huang et al., 2011).
Skin microbiota
Although less intensively studied than the gut, the skin also harbors complex communities that can have a major impact on health, and that contain both beneficial microbes and potential pathogens such as Staphylococcus aureus and Pseudomonas aeruginosa (Schommer and Gallo, 2013). The skin provides many different niches with diverse microbial populations that are subject to variable conditions and thus differ depending on the site. Skin microbial communities are shaped by factors like lifestyle, health habits and work activities. For example, communities present on the palms of the hands of 51 healthy individuals were not only very diverse, containing a large number of unique phylotypes at the species level (> 150), but were also highly variable between the right and left hands of the same individual, as well as between different individuals (Fierer et al., 2008). Differences were also found when comparing these communities with those obtained in other skin surfaces, indicating heterogeneity and lack of uniformity in the distribution of various microorganisms (Fierer et al., 2008).
Bacterial communities are also influenced by ethnicity, lifestyle and / or geographic location. Blaser et al. (2013) compared the skin bacterial communities from Amerindians in the Venezuelan Amazon with those in healthy residents of New York (US). They found 20 bacterial phyla, three of which were predominant in all samples (Firmicutes, Proteobacteria and Actinobacteria), but differences at the genus level: US residents contained predominantly Propionibacterium, Staphylococcus predominated in one of two Amerindian clusters, while the second Amerindian cluster had no single dominant taxon and a wide range of Proteobacteria (Blaser et al., 2013).
Respiratory tract microbiota
The respiratory tract is the main portal of entry for microbes such as viruses, bacteria and fungi that can colonize and in some cases trigger disease (Delhaes et al., 2012). As with other body locations, the microbial communities in different segments of the respiratory tract are shaped based on tissue type and niche characteristics. The upper respiratory tract extends from the nasal passages, through sinuses, pharynx (divided into nasopharynx and oropharynx), larynx and trachea, while the lower respiratory tract, begins at the terminal portion of the trachea and branches into the bronchi and lungs, and finally the alveoli, where gas exchange occurs.
Culture-based studies have shown differences among these anatomical sites, reflecting niche-specific conditions that select for specific communities. Bacteria of the genera Corynebacterium, Propionibacterium and Staphylococcus, including the pathogenic species S. aureus, are characteristic of the nostrils. The oropharynx (located behind the mouth) contains species belonging to the genera Streptococcus, Haemophilus, Neisseria, and to a lesser extent Staphylococcus, as well as anaerobic bacteria. The oropharynx is also epidemiologically important for carrier status of microorganisms that are pathogenic to humans: S. pneumoniae, S. pyogenes, Haemophilus influenzae, Neisseria meningitidis, Moraxella catarrhalis and S. aureus, among others (Mertz et al., 2007). Culture-independent studies have revealed greater diversity but also support differences based on niche characteristics, with limited number of taxa common to all sites and a small degree of conservation between individuals (Dethlefsen et al., 2007). The nose and oropharynx are found to have an inverse correlation between bacterial groups at each site that could be important for maintenance of a healthy microbiota (Lemon et al., 2010). In addition, the microbiota of the oropharynx is reported to be more diverse than that of the nasal cavity, although with less variation among individuals (Lemon et al., 2010; Botero et al., 2014).
The upper respiratory tract, just as the skin, is sensitive to external factors and therefore to selection of certain microbial communities. For example, cigarette smoking influences the colonization of the upper respiratory tract by certain pathogens. Both active and passive smokers have a higher risk of carrying pathogenic organisms in their airways. Cigarette smoking can promote colonization by bacteria that attach easily to oral epithelial cells, altering the mucociliary system or the host immune response against pathogens. Cigarettes themselves can also contain a wide range of potential pathogens, such as Acinetobacter, Bacillus, Burkholderia, Clostridium, Klebsiella, P. aeruginosa and Serratia, becoming a direct source of exposure to disease-causing organisms (Sapkota et al., 2010). In the nasopharynx (upper part of throat behind the nose), smokers harbor fewer microorganisms capable of interfering with colonization of pathogens, and have more microbial groups capable of producing disease than nonsmokers (Brook and Gober, 2005).
Microbiota of the lungs
The study of the lung microbiome is gaining importance in terms of its possible role in relation to respiratory disease. Although the lungs of healthy individuals were considered sterile based on results of traditional culture-based analysis, recent culture-independent studies have shown that healthy lungs are colonized by diverse bacteria (Hilty et al., 2010; Charlson et al., 2011; Erb-Downward et al., 2011; Huang et al., 2011; Segal and Blaser, 2014), and that many of these come from the oral cavity (Bassis et al., 2015). The most commonly identified phyla are Proteobacteria, Firmicutes and Bacteroidetes, even though published results tend to differ. A few potential pathogens, such as Haemophilus and Neisseria have also been detected (Beck et al., 2012).
Most of the research on lung microbiota has been focused on diseases characterized by chronic inflammation of the airways and lungs, such as cystic fibrosis (CF) (Delhaes et al., 2012; Filkins et al., 2012; Goddard et al., 2012; Willner et al., 2012), chronic obstructive pulmonary disease (COPD) (Zhang et al., 2010; Cabrera-Rubio et al., 2012), and asthma (Hilty et al., 2010). The microbiota in other respiratory conditions, such as community-acquired pneumonia (Zhou et al., 2010), the influenza H1N1 pandemic (Chaban et al., 2013) and pulmonary tuberculosis (Cui et al., 2012, Cheung et al., 2013, Botero et al., 2014) are also being studied in order to elucidate its effect on infection or disease outcome. Pulmonary CF has been traditionally associated with the infection of known pathogens such as P. aeruginosa, S. aureus and Burkholderia cepacia. However, CF patients appear to have a complex microbial ecology in their lungs. Guss et al. (2011), for example, determined the presence of over 60 bacterial genera in the sputum of four patients by using a combination of cloning, culture and pyrosequencing.
Some studies have reported changes in the lung microbiota that correlate with the health status of an individual. For example, Haemophilus species were more prevalent in the bronchoalveolar lavage of patients with COPD than in healthy controls, while Bacteroidetes were more prevalent in controls (Hilty et al., 2010). Exacerbations, or worsening of COPD symptoms, have also been associated with acquisition of new strains of H. influenzae, Moraxella catarrhalis, S. pneumoniae or P. aeruginosa and members of the Enterobacteriaceae family. These findings suggest that acquisition of these strains could potentially contribute to pathogenesis of the disease or to its acute exacerbation (Sethi et al., 2002).
The relationship between the respiratory microbiota and asthma has been proposed for many years, especially since the incidence of allergic airway disease in industrialized countries has increased significantly over the past three decades (Marri et al., 2013). This observation has been linked to the "hygiene hypothesis", which says that modern medical and health practices prevent exposure to microorganisms that are essential for correct immunological development (Salvucci, 2013; Segal and Blaser, 2014). Alterations in the composition of the intestinal microbiota, as consequence of antibiotic use, changes in diet or in habits, can result in loss of microbial members that are essential for early microbial stimulation of the immune system and development of immunological tolerance (Huffnagle, 2010; Belkaid and Hand, 2014). A study carried out in 11 patients with asthma, five patients with COPD and eight controls found that some pathogenic bacteria were more frequent in the bronchi of individuals with asthma or COPD than in healthy controls, while other bacterial phyla were more common in controls (Hilty et al., 2010).
Respiratory tract microbiota and tuberculosis
The complex interaction between the human host and Mycobacterium tuberculosis the causal agent of tuberculosis (TB), as well as the resulting infection indicates that the development of TB can be a multifactorial process (Ocejo-Vinyals et al., 2012; Sharma and Mohan, 2013). Local immune responses coupled to respiratory tract microbiota can therefore determine whether the tubercle bacilli are removed or if they develop an acute or latent infection.
Two recent studies have evaluated the microbiota in sputum samples from patients with TB and healthy individuals. In one case, the microbiota was more diverse in patient sputum samples than in that secretion samples from healthy participants, with 24 phyla present in TB patients versus 17 found in healthy participants. Some bacteria were found only in TB patients, such as Stenotrophomonas, Cupriavidus, Pseudomonas, Thermus, Sphingomonas, Methylobacterium, Diaphorobacter, Comamonas and Mobilococcus tuberculosis, bacteria that may have an important role in the onset or development of pulmonary TB (Cui et al., 2012). In a second study, some phyla were predominant in sputum samples from healthy controls and TB patients (Firmicutes, Proteobacteria, Bacteroidetes, Actinobacteria and Fusobacteria), and again some groups (Proteobacteria and Bacteroidetes, as well as 16 genera) were more frequently found either in samples from TB or in controls (Firmicutes). This study suggests that there might be a core group of microorganisms common to patients with TB, although overall no differences were found in terms of diversity between samples from patients and controls (Cheung et al., 2013).
In a more recent study of the microbiota present in patients with pulmonary TB, three different types of samples (nasopharynx, oropharynx and sputum) were studied for their usefulness in assessing changes in respiratory tract bacterial and fungal communities via 16S rRNA gene and ITS1 region analysis. There was predominance of certain phyla in all samples, with differences in diversity indexes, in relative abundances and in specific taxa associated with each sample type. The only difference between patient and control microbiota was found in oropharynx samples for both bacteria and fungi. More importantly, the bacterial and fungal community structures in oropharynx and sputum samples were similar to one another, and both differed from nasal samples. The fact that the oropharynx communities varied with respect to health status, and that they resembled those in sputum samples, suggested that oropharynx samples could be used to further analyze community structure alterations associated with tuberculosis (Botero et al., 2014).
These studies indicate that the microbiota differs between patients and healthy controls, and that these differences may play an important role in the outcome of infection and the development of pulmonary TB (Cui et al., 2012; Cheung et al., 2013; Botero et al., 2014). However, further studies are needed to fully understand if the observed differences among these communities affect or are a consequence of colonization or disease. In fact, establishing relevant correlations between the microbiota or the presence of particular microbial members and a state of health or disease is difficult. Studies that could capture samples over time, especially previous to disease onset, might provide further insight into the role of the microbiota and human diseases such as TB.
CONCLUSIONS AND PERSPECTIVES
Microorganisms are involved in more than just transient interactions or pathogenicity with the human host, a fact that is slowly becoming recognized as we learn more about the human microbiota. Initial characterization of the taxonomic profiles of microorganisms that inhabit diverse body sites indicated that these communities were both diverse and abundant and that they varied depending on the niche being analyzed. More recent work has focused on understanding how these communities change during infection and disease and whether imbalances can be associated with particular health manifestations. Microorganisms of the microbiota are now considered important for the continued health of an individual and future challenges will aim to identify particular mechanisms by which these communities actually affect host health status. Future investigations are aimed at identifying not only community composition and fluctuations, but also microbial functionality with the goal of understanding how the microbiota contributes to host well being. It will be interesting to learn more about how community members interact with one another and with the host and if alterations in community composition are consequence or cause of either health or disease status. The real challenge lies not only in uncovering associations but also in learning how to harness this information to modulate the host and promote health and well-being. The future therefore promises more insightful and eye-opening research in the field of the human microbiota that will hopefully lead to a more comprehensive appreciation of the nuances involved in establishment and maintenance of this essential microbial complement.
ACKNOWLEGMENTS
This review was done as part of the compilation of literature and analysis that supported the project "Microbiota asociada al tracto respiratorio de pacientes con tuberculosis" funded by Colciencias with Grant No. 657049326148, Corporación CorpoGen and Corporación para Investigaciones Biológicas. We would like to thank Juan Manuel Anzola, Jose Ricardo Bustos (Bioinformatics Group - Corporación CorpoGen), and Jaime Robledo (Unidad de Bacteriología y Micobacterias (Corporación para Investigaciones Biológicas) for their valuable support and suggestions in the development of this work.
REFERENCES
Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, et al. Enterotypes of the human gut microbiome. Nature. 2011;473(7346):174-180. Doi: 10.1038/nature09944. [ Links ]
Bassis CM, Erb-Downward JR, Dickson RP, Freeman CM, Schmidt TM, Young VB, et al. Analysis of the upper respiratory tract microbiotas as the source of the lung and gastric microbiotas in healthy individuals. mBio. 2015;6(2):e00037. Doi: 10.1128/mBio.00037-15. [ Links ]
Beck JM, Young VB, Huffnagle GB. The microbiome of the lung. Transl Res. 2012;160(4):258-266. Doi: 10.1016/j.trsl.2012.02.005. [ Links ]
Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014;157(1):121-141. Doi: 10.1016/j.cell.2014.03.011. [ Links ]
Bik EM, Long CD, Armitage GC, Loomer P, Emerson J, Mongodin EF, et al. Bacterial diversity in the oral cavity of 10 healthy individuals. ISME J. 2010;4(8):962-974. Doi: 10.1038/ismej.2010.30. [ Links ]
Blaser MJ, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Estrada I, et al. Distinct cutaneous bacterial assemblages in a sampling of South American Amerindians and US residents. ISME J. 2013;7(1):85-95. Doi: 10.1038/ismej.2012.81. [ Links ]
Botero LE, Delgado-Serrano L, Cepeda ML, Bustos JR, Anzola JM, Del Portillo P, et al. Respiratory tract clinical sample selection for microbiota analysis in patients with pulmonary tuberculosis. Microbiome. 2014;2:29. Doi: 10.1186/2049-2618-2-29. [ Links ]
Brook I, Gober AE. Recovery of potential pathogens and interfering bacteria in the nasopharynx of smokers and nonsmokers. Chest. 2005;127(6):2072-2075. Doi: 10.1378/chest.127.6.2072. [ Links ]
Brotman RM, Bradford LL, Conrad M, Gajer P, Ault K, Peralta L, et al. Association between Trichomonas vaginalis and vaginal bacterial community composition among reproductive-age women. Sex Transm Dis. 2012;39(10):807-812. Doi: 10.1097/OLQ.0b013e3182631c79. [ Links ]
Cabrera-Rubio R, Garcia-Nunez M, Seto L, Anto JM, Moya A, Monso E, et al. Microbiome diversity in the bronchial tracts of patients with chronic obstructive pulmonary disease. J Clin Microbiol. 2012;50(11):3562-3568. Doi: 10.1128/JCM.00767-12. [ Links ]
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335-336. Doi: 10.1038/nmeth.f.303. [ Links ]
Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative view. Cell. 2012;148(6):1258-1270. Doi: 10.1016/j.cell.2012.01.035. [ Links ]
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37(suppl 1):D141-D145. Doi: 10.1093/nar/gkn879. [ Links ]
Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, et al. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014;42(D1):D633-D642. Doi: 10.1093/nar/gkt1244. [ Links ]
Costello EK, Stagaman K, Dethlefsen L, Bohannan BJ, Relman DA. The application of ecological theory toward an understanding of the human microbiome. Science. 2012;336(6086):1255-1262. Doi: 10.1126/science.1224203. [ Links ]
Cox LM, Blaser MJ. Antibiotics in early life and obesity. Nat Rev Endocrinol. 2015;11:182-190. Doi: 10.1038/nrendo.2014.210. [ Links ]
Cui Z, Zhou Y, Li H, Zhang Y, Zhang S, Tang S, et al. Complex sputum microbial composition in patients with pulmonary tuberculosis. BMC Microbiol. 2012;12:276. Doi: 10.1186/1471-2180-12-276. [ Links ]
Chaban B, Albert A, Links MG, Gardy J, Tang P, Hill JE. Characterization of the upper respiratory tract microbiomes of patients with pandemic H1N1 influenza. PLoS One. 2013;8(7):e69559. Doi: 10.1371/journal.pone.0069559. [ Links ]
Charlson ES, Bittinger K, Haas AR, Fitzgerald AS, Frank I, Yadav A, et al. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med. 2011;184(8):957-63. Doi: 10.1164/rccm.201104-0655OC. [ Links ]
Charlson ES, Chen J, Custers-Allen R, Bittinger K, Li H, Sinha R, et al. Disordered microbial communities in the upper respiratory tract of cigarette smokers. PLoS One. 2010;5(12):e15216. Doi: 10.1371/journal.pone.0015216. [ Links ]
Chen H, Jiang W. Application of high-throughput sequencing in understanding human oral microbiome related with health and disease. Front Microbiol. 2014;5:508. Doi: 10.3389/fmicb.2014.00508. [ Links ]
Cheung MK, Lam WY, Fung WY, Law PT, Au CH, Nong W, et al. Sputum microbiota in tuberculosis as revealed by 16S rRNA pyrosequencing. PLoS One. 2013;8(1):e54574. Doi: 10.1371/journal.pone.0054574. [ Links ]
Delhaes L, Monchy S, Frealle E, Hubans C, Salleron J, Leroy S, et al. The airway microbiota in cystic fibrosis: a complex fungal and bacterial community-implications for therapeutic management. PLoS One. 2012;7(4):e36313. Doi: 10.1371/journal.pone.0036313. [ Links ]
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72(7):5069-5072. Doi: 10.1128/AEM.03006-05. [ Links ]
Dethlefsen L, McFall-Ngai M, Relman DA. An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature. 2007;449(7164):811-818. Doi: 10.1038/nature06245. [ Links ]
Dore J, Blottiere H. The influence of diet on the gut microbiota and its consequences for health. Curr Opin Biotechy. 2015;32:195-199. Doi: 10.1016/j.copbio.2015.01.002. [ Links ]
Erb-Downward JR, Thompson DL, Han MK, Freeman CM, McCloskey L, Schmidt LA, et al. Analysis of the lung microbiome in the "healthy" smoker and in COPD. PLoS One. 2011;6(2):e16384. Doi:10.1371/journal.pone.0016384. [ Links ]
Fierer N, Hamady M, Lauber CL, Knight R. The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc Natl Acad Sci USA. 2008;105(46):17994-17999. Doi: 10.1073/pnas.0807920105. [ Links ]
Filkins LM, Hampton TH, Gifford AH, Gross MJ, Hogan DA, Sogin ML, et al. Prevalence of streptococci and increased polymicrobial diversity associated with cystic fibrosis patient stability. J Bacteriol. 2012;194(17):4709-4717. Doi: 10.1128/JB.00566-12. [ Links ]
Fredricks DN. Molecular methods to describe the spectrum and dynamics of the vaginal microbiota. Anaerobe. 2011;17(4):191-195. Doi: 10.1016/j.anaerobe.2011.01.001. [ Links ]
Gevers D, Cohan FM, Lawrence JG, Spratt BG, Coenye T, Feil EJ, et al. Opinion: Re-evaluating prokaryotic species. Nat Rev Microbiol. 2005;3(9):733-739. Doi: 10.1038/nrmicro1236. [ Links ]
Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, et al. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312(5778):1355-1359. Doi: 10.1126/science.1124234. [ Links ]
Glendinning L, Free A. Supra-Organismal Interactions in the Human Intestine Front Cell Infect Microbiol. 2014;4(47):1-4. Doi:10.3389/fcimb.2014.00047. [ Links ]
Goddard AF, Staudinger BJ, Dowd SE, Joshi-Datar A, Wolcott RD, Aitken ML, et al. Direct sampling of cystic fibrosis lungs indicates that DNA-based analyses of upper-airway specimens can misrepresent lung microbiota. Proc Natl Acad Sci USA. 2012;109(34):13769-13774. Doi: 10.1073/pnas.1107435109. [ Links ]
Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, et al. Topographical and temporal diversity of the human skin microbiome. Science. 2009;324(5931):1190-1192. Doi: 10.1126/science.1171700. [ Links ]
Gross EL, Leys EJ, Gasparovich SR, Firestone ND, Schwartzbaum JA, Janies DA, et al. Bacterial 16S sequence analysis of severe caries in young permanent teeth. J Clin Microbiol. 2010;48(11):4121-4128. Doi: 10.1128/JCM.01232-10. [ Links ]
Group NHW, Peterson J, Garges S, Giovanni M, McInnes P, Wang L, et al. The NIH Human Microbiome Project. Genome Res. 2009;19(12):2317-2323. Doi:10.1101/gr.096651.109. [ Links ]
Guss AM, Roeselers G, Newton IL, Young CR, Klepac-Ceraj V, Lory S, et al. Phylogenetic and metabolic diversity of bacteria associated with cystic fibrosis. ISME J. 2011;5(1):20-29. Doi: 10.1038/ismej.2010.88. [ Links ]
Hamady M, Knight R. Microbial community profiling for human microbiome projects: Tools, techniques, and challenges. Genome Res. 2009;19(7):1141-1152. Doi: 10.1101/gr.085464.108. [ Links ]
Handelsman J. Metagenomics: application of genomics to uncultured microorganisms. Microbiol Mol Biol Rev. 2004;68(4):669-685. Doi: 10.1128/MMBR.68.4.669-685.2004. [ Links ]
Hartman AL, Riddle S, McPhillips T, Ludascher B, Eisen JA. Introducing W.A.T.E.R.S.: a workflow for the alignment, taxonomy, and ecology of ribosomal sequences. BMC bioinformatics. 2010;11:317. Doi: 10.1186/1471-2105-11-317. [ Links ]
Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, et al. Disordered microbial communities in asthmatic airways. PLoS One. 2010;5(1):e8578. Doi: 10.1371/journal.pone.0008578. [ Links ]
Huang S, Yang F, Zeng X, Chen J, Li R, Wen T, et al. Preliminary characterization of the oral microbiota of Chinese adults with and without gingivitis. BMC Oral Health. 2011;11:33. Doi: 10.1016/j.jaci.2010.10.048. [ Links ]
Huang YJ, Nelson CE, Brodie EL, Desantis TZ, Baek MS, Liu J, et al. Airway microbiota and bronchial hyperresponsiveness in patients with suboptimally controlled asthma. J Allergy Clin Immunol. 2011;127(2):372-381. Doi: 10.1016/j.jaci.2010.10.048. [ Links ]
Huffnagle GB. The microbiota and allergies/asthma. PLoS Pathog. 2010;6(5):e1000549. Doi: 10.1371/journal.ppat.1000549. [ Links ]
Kim M, Oh HS, Park SC, Chun J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int J Syst Evol Microbiol. 2014;64(2):346-351. Doi:10.1099/ijs.0.059774-0. [ Links ]
Lee S, Sung J, Lee J, Ko G. Comparison of the gut microbiotas of healthy adult twins living in South Korea and the United States. Appl Environ Microbiol. 2011;77(20):7433-7437. Doi: 10.1128/AEM.05490-11. [ Links ]
Lemon KP, Armitage GC, Relman DA, Fischbach MA. Microbiota-targeted therapies: an ecological perspective. Sci Transl Med. 2012;4(137):137rv5. Doi: 10.1126/scitranslmed.3004183. [ Links ]
Lemon KP, Klepac-Ceraj V, Schiffer HK, Brodie EL, Lynch SV, Kolter R. Comparative analyses of the bacterial microbiota of the human nostril and oropharynx. MBio. 2010;1(3):e00129-10. Doi: 10.1128/mBio.00129-10. [ Links ]
Ley RE, Lozupone CA, Hamady M, Knight R, Gordon JI. Worlds within worlds: evolution of the vertebrate gut microbiota. Nat Rev Microbiol. 2008;6(10):776-788. Doi: 10.1038/nrmicro1978. [ Links ]
Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124(4):837-848. Doi: 10.1016/j.cell.2006.02.017. [ Links ]
Ling Z, Kong J, Liu F, Zhu H, Chen X, Wang Y, et al. Molecular analysis of the diversity of vaginal microbiota associated with bacterial vaginosis. BMC Genomics. 2010;11:488. Doi: 10.1186/1471-2164-11-488. [ Links ]
Ma B, Forney LJ, Ravel J. Vaginal microbiome: rethinking health and disease. Annu Rev Microbiol. 2012;66:371-389. Doi: 10.1146/annurev-micro-092611-150157. [ Links ]
Marri PR, Stern DA, Wright AL, Billheimer D, Martinez FD. Asthma-associated differences in microbial composition of induced sputum. J Allergy Clin Immunol. 2013;131(2):346-352. Doi: 10.1016/j.jaci.2012.11.013. [ Links ]
Mertz D, Frei R, Jaussi B, Tietz A, Stebler C, Fluckiger U, et al. Throat swabs are necessary to reliably detect carriers of Staphylococcus aureus. Clin Infect Dis. 2007;45(4):475-477. Doi: 10.1086/520016. [ Links ]
Morgan XC, Huttenhower C. Chapter 12: Human microbiome analysis. PLoS Comput Biol. 2012;8(12):e1002808. Doi: 10.1371/journal.pcbi.1002808. [ Links ]
Muegge BD, Kuczynski J, Knights D, Clemente JC, Gonzalez A, Fontana L, et al. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science. 2011;332(6032):970-974. Doi:10.1126/science.1198719. [ Links ]
Mueller NT, Bakacs E, Combellick J, Grigoryan Z, Dominguez-Bello MG. The infant microbiome development: mom matters. Trends in molecular medicine. 2015;21(2):109-117. Doi:10.1038/ncomms7342. [ Links ]
O'Keefe SJ, Li JV, Lahti L, Ou J, Carbonero F, Mohammed K, et al. Fat, fibre and cancer risk in African Americans and rural Africans. Nature communications. 2015;6:6342. Doi: 10.1038/ncomms7342. [ Links ]
Ocejo-Vinyals JG, Lavin-Alconero L, Sanchez-Velasco P, Guerrero-Alonso MA, Ausin F, Farinas MC, et al. Mannose-binding lectin promoter polymorphisms and gene variants in pulmonary tuberculosis patients from cantabria (northern Spain). Pulm Med. 2012;(2012):1-6. Doi: 10.1155/2012/469128. [ Links ]
Proctor LM. The Human Microbiome Project in 2011 and beyond. Cell Host Microbe. 2011;10(4):287-291. Doi: 10.1016/j.chom.2011.10.001. [ Links ]
Ravel J, Gajer P, Abdo Z, Schneider GM, Koenig SS, McCulle SL, et al. Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci USA. 2011;108(Suppl1):4680-4687. Doi: 10.1073/pnas.1002611107. [ Links ]
Salvucci E. El Agotamiento del bioma y sus consecuencias. Acta biol Colomb. 2013;18(1):31-42. [ Links ]
Sapkota AR, Berger S, Vogel TM. Human pathogens abundant in the bacterial metagenome of cigarettes. Environ Health Perspect. 2010;118(3):351-356. Doi: 10.1128/AEM.01541-09. [ Links ]
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75(23):7537-7541. Doi: 10.1128/AEM.01541-09. [ Links ]
Schommer NN, Gallo RL. Structure and function of the human skin microbiome. Trends Microbiol. 2013;21(12):660-668. Doi: 10.1016/j.tim.2013.10.001. [ Links ]
Segal LN, Blaser MJ. A brave new world: the lung microbiota in an era of change. Anna Am Thorac Soc. 2014;11(Suppl1):S21-S27. Doi:10.1513/AnnalsATS.201306-189MG. [ Links ]
Sethi S, Evans N, Grant BJ, Murphy TF. New strains of bacteria and exacerbations of chronic obstructive pulmonary disease. N Engl J Med. 2002;347(7):465-471. Doi: 10.1056/NEJMoa012561. [ Links ]
Sharma R, Young C, Neu J. Molecular modulation of intestinal epithelial barrier: contribution of microbiota. J Biomed Biotechnol. 2010;2010:1-15. Doi: 10.1155/2010/305879. [ Links ]
Sharma SK, Mohan A. Tuberculosis: From an incurable scourge to a curable disease-journey over a millennium. Indian J Med Res. 2013;137(3):455-493. Doi: IndianJMedRes_2013_137_3_455_110999. [ Links ]
Silva MJ, Carneiro MB, dos Anjos Pultz B, Pereira Silva D, Lopes ME, dos Santos LM. The multifaceted role of commensal microbiota in homeostasis and gastrointestinal diseases. J Immunol Res. 2015;2015:1-14. Doi: 10.1155/2015/321241. [ Links ]
Stearns JC, Lynch MD, Senadheera DB, Tenenbaum HC, Goldberg MB, Cvitkovitch DG, et al. Bacterial biogeography of the human digestive tract. Sci Rep. 2011;1:170. Doi: 10.1038/srep00170. [ Links ]
Suez J, Korem T, Zeevi D, Zilberman-Schapira G, Thaiss CA, Maza O, et al. Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature. 2014;514(7521):181-186. Doi: 10.1038/nature13793. [ Links ]
Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457(7228):480-484. Doi:10.1038/nature07540. [ Links ]
Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. 2007;449(7164):804-810.Doi:10.1038/nature06244. [ Links ]
Ursell LK, Clemente JC, Rideout JR, Gevers D, Caporaso JG, Knight R. The interpersonal and intrapersonal diversity of human-associated microbiota in key body sites. J Allergy Clin Immunol. 2012;129(5):1204-1208. Doi:10.1016/j.jaci.2012.03.010. [ Links ]
Wang WL, Xu SY, Ren ZG, Tao L, Jiang JW, Zheng SS. Application of metagenomics in the human gut microbiome. World J Gastroenterol. 2015;21(3):803-814. Doi:10.3748/wjg.v21.i3.803. [ Links ]
Willner D, Haynes MR, Furlan M, Schmieder R, Lim YW, Rainey PB, et al. Spatial distribution of microbial communities in the cystic fibrosis lung. ISME J. 2012;6(2):471-474. Doi:10.1038/ismej.2011.104. [ Links ]
Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334(6052):105-108. Doi:10.1126/science.1208344. [ Links ]
Zaneveld J, Turnbaugh PJ, Lozupone C, Ley RE, Hamady M, Gordon JI, et al. Host-bacterial coevolution and the search for new drug targets. Curr Opin Chem Biol. 2008;12(1):109-114. Doi: 10.1016/j.cbpa.2008.01.015. [ Links ]
Zhang M, Li Q, Zhang XY, Ding X, Zhu D, Zhou X. Relevance of lower airway bacterial colonization, airway inflammation, and pulmonary function in the stable stage of chronic obstructive pulmonary disease. Eur J Clin Microbiol Infect Dis. 2010;29(12):1487-1493. Doi: 10.1007/s10096-010-1027-7. [ Links ]
Zhou X, Brown CJ, Abdo Z, Davis CC, Hansmann MA, Joyce P, et al. Differences in the composition of vaginal microbial communities found in healthy Caucasian and black women. ISME J. 2007;1(2):121-133. Doi:10.1038/ismej.2007.12. [ Links ]
Zhou Y, Lin P, Li Q, Han L, Zheng H, Wei Y, et al. Analysis of the microbiota of sputum samples from patients with lower respiratory tract infections. Acta Biochim Biophys Sin. 2010;42(10):754-761. Doi:10.1093/abbs/gmq081. [ Links ]
