










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Biológica Colombiana
Print version ISSN 0120-548X
Acta biol.Colomb. vol.21 no.3 Bogotá Sept./Dec. 2016
https://doi.org/10.15446/abc.v21n3.54712
DOI: http://dx.doi.org/10.15446/abc.v21n3.54712
CARACTERIZACIÓN MOLECULAR DEL Potato virus V (PVV) INFECTANDO Solanum phureja MEDIANTE SECUENCIACIÓN DE NUEVA GENERACIÓN
Molecular Characterization of Potato virus V (PVV) Infecting Solanum phureja Using Next-Generation Sequencing
Daniela ÁLVAREZ YEPES1; Pablo Andrés GUTIÉRREZ SÁNCHEZ1; Mauricio MARÍN MONTOYA1.
1 Laboratorios de Microbiología Industrial y Biología Celular y Molecular, Facultad de Ciencias, Universidad Nacional de Colombia, Sede Medellín. Calle 59A n°. 63-20, bloque 19A. Medellín, Colombia.
For correspondence. mamarinm@unal.edu.co
Received: 16th December 2015, Returned for revision: 28th February 2016, Accepted: 20th April 2016.
Associate Editor: Leonardo Galindo.
Citation/Citar este artículo como: Álvarez Yepes D, Gutiérrez Sánchez PA, Marín Montoya M. Caracterización molecular del Potato virus V (PVV) infectando Solanum phureja mediante secuenciación de nueva generación. Acta biol. Colomb. 2016;21(3):521-531. DOI: http://dx.doi.org/10.15446/abc.v21n3.54712
RESUMEN
Las enfermedades virales son uno de los problemas más limitantes para la producción de papa en el mundo. Uno de los materiales de papa más susceptibles a los virus corresponde a Solanum phureja; sin embargo, en Colombia son pocos los estudios adelantados sobre los agentes causales que lo afectan. En este trabajo se realizó una caracterización molecular del Potato virus V (PVV) infectando plantas de S. phureja en Antioquia, utilizando métodos de secuenciación de nueva generación (NGS), pruebas de DAS-ELISA, RT-PCR en tiempo real (RT-qPCR) y RT-PCR convencional. Los resultados indican la ocurrencia de niveles muy variables de incidencia del virus entre lotes de cultivo (6,7 % a 86 %). El PVV tiene un genoma de 9828 nt que codifica para una poliproteína de 3066 aa y presenta dos variantes principales (Var_A y Var_B) en proporciones de 72 y 28 %. Estas variantes comparten altos niveles de identidad genética (99,7 % en todo el genoma) entre ellas y con respecto a la cepa PVV-Phureja reportada en Colombia, pero no con otras cepas del mundo (82-83 %). Con base en dichos genomas, se diseñaron y evaluaron en muestras foliares de S. phureja, dos pares de cebadores para la detección del virus en pruebas de RT-PCR (459 pb) y RT-qPCR (89 pb, Ct=12,08-21,86 y Tm= 78,7°C-80,2 °C), confirmándose la presencia de este virus en tejidos sintomáticos y asintomáticos de papa criolla. La ocurrencia generalizada de PVV en los cultivos de S. phureja indica la necesidad de incorporar en los programas de certificación de tubérculos-semilla de S. phureja en Colombia el diagnóstico de este virus.
Palabras clave: DAS-ELISA, NGS, papa, Potyvirus, RT-qPCR.
ABSTRACT
Viral diseases are one of the most limiting problems in the production of potato worldwide. Solanum phureja constitutes one of the most susceptible materials to viral diseases in Colombia; however, there are few studies on viruses infecting this crop. In the current study, we performed a molecular characterization of Potato virus V (PVV) that infects S. phureja, using different potato plots located in the province of Antioquia, using Next-Generation Sequencing (NGS), DAS-ELISA, real time RT-PCR (RT-qPCR) and RT-PCR. Results revealed variable levels of incidence among plots (6.7 %-86 %) and the presence of two slightly different variants (Var_A and Var_B) present in approximately 72 %:28 % ratio. These PVV strains have a genome of 9828 nt codifying for a polyprotein of 3066 aa and share high nucleotide sequence identity (99,7 % in their complete genome) with respect to PVV-Phureja, recently described in Colombia, but are very divergent with respect to currently available PVV genomes (82-83 %). The genome information was used to design two sets of primers, useful in the specific detection of this virus in S. phureja leaf samples through RT-PCR (459 bp) and RT-qPCR (89 bp, Ct=12.08-21.86; Tm=78.7 °C-80.2 °C). This study underscores the importance of including diagnostics of PVV in S. phureja tuber-seed certification programs in Colombia.
Keywords: DAS-ELISA, NGS, potato, Potyvirus, RT-qPCR.
INTRODUCCIÓN
Solanum tuberosum L. subsp. andigena Hawkes y Solanum phureja Juz. & Bukasov, son las dos principales especies de papa cultivadas en Colombia. Aunque de las 160000 ha que se siembran con papa en el país, solo cerca del 10 % corresponde a variedades de S. phureja, este cultivo ha recibido gran atención en los últimos años, dada su alta demanda interna y gran potencial de exportación debido a sus excelentes características organolépticas, altos niveles de vitaminas B y C y de minerales (Fe, Zn, Cu y Ca) (Rodríguez et al., 2013).
Solanum phureja es una especie diploide de papa que produce tubérculos de color amarillo y presenta como centro de origen los Andes suramericanos, siendo el suroccidente de Colombia uno de sus mayores centros de biodiversidad (Rodríguez et al., 2013). Esta especie presenta ciclos cortos de cultivo (cuatro a cinco meses) y ausencia de dormancia en los tubérculos. Entre los cultivares más sembrados actualmente en el país se destacan Criolla Colombia, Criolla Latina y Criolla Paisa (Rodríguez et al., 2009; Gúzman-Barney et al., 2012). En Colombia se obtiene una producción anual de S. phureja entre 100000 y 150000 t con rendimientos promedio de siete a 12 t/ha, inferiores a su óptimo agronómico (15 a 25 t/ha), como resultado de los bajos niveles de tecnificación del cultivo, especialmente en lo referente a la calidad del tubérculo-semilla y al manejo de plagas y enfermedades (Gúzman-Barney et al., 2012).
Las enfermedades virales son uno de los problemas fitopatológicos más limitantes para la producción de S. phureja (Gúzman-Barney et al., 2012; García et al., 2016; Gutiérrez et al., 2016), por ejemplo, se ha reportado que en este cultivo la infección del Potato yellow vein virus (PYVV) puede reducir sus rendimientos en niveles del 40 % al 50 % (Gúzman-Barney et al., 2012). Además del PYVV, otros virus detectados con altos niveles de incidencia en S. phureja incluyen el Potato virus Y (PVY), Potato virus S (PVS), Potato virus X (PVX) y Potato leafroll virus (PLRV) (Gil et al., 2013; Gutiérrez et al., 2013; Hernández-Guzmán y Gúzman-Barney, 2014; Medina et al., 2015; García et al., 2016).
Recientemente, utilizando metodologías de secuenciación de nueva generación (NGS), se encontraron plantas de S. phureja infectadas con Andean potato latent virus (APLV) (Kreuze et al., 2013) y Potato virus V (PVV) (Gutiérrez et al., 2014; Gutiérrez et al., 2016), virus cuya presencia no había sido confirmada en el país. El hallazgo de esta última especie (PVV) sobre S. phureja en Antioquia resultó inesperado por cuanto los síntomas de mosaicos que presentaban las plantas infectadas habían sido asociados con la infección por PVY. PVV ha sido reportado en Perú (Spetz et al., 2003), Costa Rica (Vásquez et al., 2006), Estados Unidos (Shiel et al., 2004), Europa continental e Inglaterra (Oruetxebarria et al., 2000; Mortensen et al., 2010) y en Irán (Shamsadden-Saeed et al., 2014).
PVV es un miembro del género Potyvirus (familia Potyviridae), que fue inicialmente considerado como una variante de PVY; sin embargo, análisis serológicos y filogenéticos confirmaron su identidad como una especie diferente (Oruetxebarria et al., 2000; Oruetxebarria et al., 2001; Spetz et al., 2003). El virus consiste de partículas flexuosas de 760 nm, cuyo material genético es ARN de cadena sencilla positiva (ssRNA+) con cerca de 9900 nt (excluyendo la cola de poli-A). Su genoma contiene un gran marco de lectura abierto (ORF) que codifica para una poliproteína de 3066-3068 residuos y presenta regiones no traducidas (UTR) de aproximadamente 200 (3'UTR) y 445 nt (5'UTR), respectivamente (Oruetxebarria et al., 2000; Shamsadden-Saeed et al., 2014; Gutiérrez et al., 2016).
Los síntomas asociados a la infección por PVV incluyen mosaicos suaves y disminución del tamaño de la lámina foliar; aunque sobre algunas variedades puede inducir mosaicos severos y manchas necróticas en hojas bajeras (Calvert et al., 1980; Jones, 1984). PVV es transmitido por tubérculos-semilla infectados y por diversos áfidos de manera no persistente (Myzus persicae, Brachycaudus helichrysi, Macrosiphum euphorbiae, etc.) (Calvert et al., 1980; Fribourg y Nakashima, 1984).
Con el fin de aumentar el nivel de conocimiento que se tiene del PVV afectando los cultivos de S. phureja del país, en este trabajo se evaluó la incidencia de este virus mediante pruebas de DAS-ELISA en cuatro cultivos en Antioquia; así como también se realizó la caracterización molecular de su genoma completo a partir del análisis del transcriptoma de tejidos foliares de plantas de S. phureja procedentes del municipio de La Unión en este departamento. Finalmente, se diseñaron dos pares de cebadores específicos para la detección de PVV por RT-PCR convencional y RT-qPCR, validándose su utilidad a partir de un grupo de muestras foliares sintomáticas y asintomáticas de esta especie de papa.
MATERIALES Y MÉTODOS
Muestras
En cuatro lotes de cultivo de S. phureja cv. Criolla Colombia, ubicados en la zona rural de los municipios de Entrerríos (Lote 1) (6° 33' 0'' Norte, 75° 31' 1'' Oeste), Marinilla (Lotes 2 y 3) (6° 10' 1'' Norte, 75° 19' 59'' Oeste) y El Peñol (Lote 4) (6° 13' 1'' Norte, 75° 13' 1'' Oeste) (Antioquia, Colombia), se colectaron 15 muestras aleatorias de hojas. Cuando se encontraban plantas con posibles síntomas asociados a virus (diferentes del amarillamiento de nervaduras causado por PYVV), se colectaron muestras de tejidos sintomáticos, siendo obtenidos siete grupos de muestras: amarillamiento (2), moteado (1), enanismo (1), enrollamiento foliar (1) y mosaico (2). Finalmente, en un lote de S. phureja en estado de floración ubicado en el municipio de La Unión (Antioquia), se obtuvo un grupo de tejidos foliares de diferentes plantas con síntomas de mosaico, que tradicionalmente han sido asociados por los técnicos y agricultores de papa con la infección por PVY, para la secuenciación de su transcriptoma por NGS.
Pruebas de DAS-ELISA
Se evaluó la presencia de PVV en las 60 muestras colectadas en los cuatro lotes de cultivo, así como en las muestras sintomáticas (incluyendo aquellas destinadas a NGS) utilizando la prueba DAS-ELISA #SRA 10100 de la compañía Agdia (Indiana, EEUU), siguiendo las instrucciones del fabricante. Los resultados colorimétricos fueron cuantificados en un equipo Multiscan RC/MS/EX (Labsystem, EEUU). En cada prueba fue incluido un control positivo (LPC 10100 "method" control positivo) y un control negativo (LNC 10100) proveídos por Agdia, consistentes en tejidos vegetales liofilizados infectados o libres de PVV, respectivamente. Se estableció como criterio de definición de pruebas positivas, aquel reportado por la compañía serológica Bioreba (BIOREBA, 2015), que utiliza como valor de corte la fórmula: Cut-off = (promedio + 3 desviaciones estándar) x 1,1.
Secuenciación de nueva generación (NGS)
La secuenciación de nueva generación del transcriptoma de S. phureja fue realizada a partir de ARN total, el cual se extrajo mediante el kit GeneJET Plant RNA Purification Mini (Thermo, EEUU), procediéndose a la degradación del ARN ribosomal (ARNr) con el sistema TruSeq Stranded Total RNA with Ribo-Zero Plant (Illumina, EEUU). Para la construcción de la librería de ADN copia (ADNc) y la unión de los adaptadores (TruSeq Index Adapters) necesarios para las reacciones de ¨PCR-bridge¨, se utilizó el kit TruSeq RNA Sample Preparation Kit (Illumina, EEUU). La secuenciación se realizó en una línea hasta obtener nueve millones de reads pareados, utilizando el equipo Illumina HiSeq 2000 en la compañía Macrogen (Corea del Sur). Una vez obtenidas las secuencias, se eliminaron aquellas de los adaptadores y las bases de baja calidad (Phred <30) utilizando el programa SeqTK (GitHub, 2015). Los reads de origen viral fueron seleccionados luego de su comparación con bases de datos locales utilizando BLASTN (Gish y States, 1993) y el genoma de PVV fue ensamblado por mapeo con el programa Bowtie2 (Langmead y Salzberg, 2012) utilizando como referencia el genoma de la cepa PVV-Phureja (gb: KP849483). Las regiones del genoma que codifican para las diez proteínas maduras de PVV fueron identificadas con BLASTX (Gish y States, 1993).
Detección de PVV por RT-PCR en tiempo real
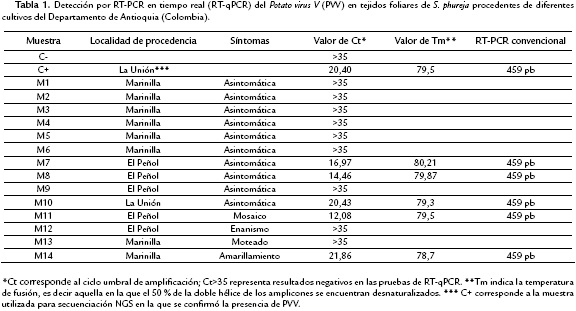
Con base en el alineamiento de los genomas de PVV obtenidos en este trabajo y en aquel reportado por Gutiérrez et al., (2016), se diseñaron un par de cebadores específicos para la detección por RT-qPCR de PVV. Para esto se empleó el programa Primer-Blast (Ye et al., 2012), siendo seleccionados los cebadores PVV_phu_F (5'-ATG CTG GAA AAG ATC CAG C-3') y qPVV_phu_R (5'-CAT CCC GCT CCT CAA C-3') que amplifican un fragmento de 89 pb de la cápside viral (CP) y se ubican entre las posiciones del genoma 8621-8639 y 8695-8710, respectivamente. La validez de los cebadores fue evaluada en 14 muestras de tejidos foliares de papa, diez correspondientes a muestras aleatorias procedentes de los lotes de S. phureja utilizados en el estudio y cuatro a muestras de tejidos con síntomas asociados a infecciones virales (amarillamiento, mosaico, moteado y enanismo) (Tabla 1). De cada una de estas muestras se extrajo el ARN con el kit GeneJET Plant RNA Purification (Thermo), siguiendo las instrucciones del fabricante. El ARN así obtenido fue eluído en 50 μL de agua tratada con DEPC y determinada su concentración y pureza por lecturas de absorbancia a 260 nm y 280 nm en un equipo Nanodrop 2000C (Thermo).
Las reacciones de RT-qPCR se realizaron en dos pasos utilizando el sistema SYBR Green I. La retrotranscripción consistió de 20 µL con 200 U de la enzima Maxima Reversa Transcriptasa (Thermo), 1X de buffer RT, 0,5 mM de dNTPs, 20 pmol del primer Oligo-(dT)18, 20 U de inhibidor de ARNasas (RiboLock, Thermo) y 50-100 ng del ARN total. Las reacciones se incubaron en un termociclador T3 (Biometra, Alemania) a 65 °C por 5 min, seguido de 50 °C por 30 min y 85 °C por 5 min para desnaturalizar la enzima. Las reacciones de qPCR se realizaron en 25 μL conteniendo 12,5 μL del kit Maxima SYBR Green/ROX qPCR Master Mix (2X) (Thermo), 10 μL de agua tratada con DEPC, 0,3 μM de los cebadores PVV_phu_F y qPVV_phu_R y 50-100 ng de ADNc. La amplificación se adelantó en un equipo Rotor-Gene Q-5plex Platform (Qiagen, Alemania) y consistió de 95 °C por 10 min para activar la Taq-polimerasa (Hot-start), seguido por 35 ciclos de 95 °C por 10 s y 52 °C por 45 s. La adquisición de fluorescencia se realizó después de cada ciclo de amplificación y los valores de ciclo umbral (Ct) para cada muestra fueron definidos utilizando los parámetros por defecto del programa Rotor-Gene Q ver. 1.7., siendo consideradas como positivas aquellas muestras que superaron el valor de Ct antes del ciclo 35. Los amplicones fueron sometidos a un análisis de desnaturalización utilizando la herramienta del equipo HRM (High Resolution Melting) entre 50 y 99 °C y comparados los valores de la temperatura de fusión (Tm) con respecto a los alcanzados para el control positivo secuenciado mediante NGS en este trabajo. Todas las reacciones incluyeron un control negativo libre de ADNc viral. Finalmente, cuatro de los amplicones correspondientes a tres muestras de tejidos de papa y al control positivo de PVV fueron secuenciados por el método de Sanger en la compañía Macrogen, previa purificación con el kit QIAquick Gel Extraction (Qiagen). Las secuencias fueron editadas con el software Mega 6.0 (Tamura et al., 2013) y comparadas con GenBank mediante BLASTN.
Detección de PVV por RT-PCR
Con base en los genomas disponibles para PVV, también se diseñó el par de cebadores PVV_phu_F y PVV_phu_R (5'-TGA AAG TGG GCT TTG CG-3') para la amplificación por RT-PCR convencional de un fragmento de 459 pb del gen CP de este virus. Las reacciones de RT-PCR se realizaron en dos pasos; la retrotranscripción a 65 °C por 5 min, 50 °C por 30 min y 85 °C por 5 min, en un volumen final de 20 µL conteniendo 100 U de Maxima Reversa transcriptasa (Thermo), 1X de buffer RT, 0,5 mM de dNTPs, 20 pmol del cebador Oligo-(dT)18, 20 U de inhibidor de ARNasas y 2,5 µL del ARN. Por su parte, la PCR se realizó en un volumen de 25 µL que incluía 17,8 µL de agua, 1X buffer de enzima (10X), 1,8 mM de MgCl2, 0,2 mM de dNTPs, 0,2 µM de los cebadores, 1 U de Taq ADN polimerasa (Thermo) y 50-100 ng de ADNc. El programa de amplificación correspondió a 95°C por 3 min, seguido de 40 ciclos de 94 °C por 30 s, 52 °C por 1 min, 72 °C por 1 min y una extensión final a 72 °C por 5 min. El tamaño de los amplicones fue determinado por electroforesis en gel de agarosa al 1,8 %, usando tinción con GelRed 1X (Biotium, EEUU), en un equipo Bio Doc Analyze (Biometra, Alemania). Finalmente, cuatro de los amplicones que presentaron el tamaño esperado fueron purificados directamente del gel utilizando el kit GeneJET Gel Extraction (Thermo) y secuenciados en ambas direcciones en un equipo ABI Prism 3730xl (PE Applied Biosystems, EEUU) de la compañía Macrogen (Corea del Sur).
Análisis filogenéticos
Las secuencias obtenidas con cada cebador fueron editadas mediante el software BioEdit 6.0.6 (Hall, 1999), construyéndose secuencias consenso y confirmándose su identidad por comparación del GenBank utilizando BLASTN. Aquellas que resultaron positivas para la región CP de PVV, fueron alineadas mediante el software Clustal W (Larkin et al., 2007) con secuencias de este gen reportadas para aislamientos de PVV de diferentes países, así como de otros potyvirus de referencia utilizados como grupos externos de análisis [Pepper yellow mosaic virus (PepYMV), Peru tomato virus (PTV), Ecuadorian rocoto virus (ERV), Brugmansia suaveolens mottle virus (BsMoV), Wild potato mosaic virus (WPMV)]. Posteriormente, se realizó un análisis filogenético con dichas secuencias de CP haciendo uso del software Mega 6.0 (Tamura et al., 2013) con los algoritmos Neighbor-Joining con el modelo Kimura 2-parámetros y Máxima verosimilitud con el modelo Jukes-Cantor. El soporte de la topología interna de las ramas de los dendrogramas fue determinado por análisis de bootstrap con 1000 remuestreos (Felsenstein, 1985). En todos los análisis realizados se incluyó como base de comparación, la región correspondiente a los genomas de PVV obtenidos en el presente estudio y en aquel de Gutiérrez et al., (2016). Adicionalmente, se realizaron análisis filogenéticos con las secuencias codificantes para la poliproteína de dichos genomas de PVV y de los otros potyvirus de referencia. Los números de accesión del GenBank para las secuencias de las dos variantes de PVV obtenidas en este trabajo corresponden a KT985458 (PVV_VarA) y KT985459 (PVV_VarB).
RESULTADOS
Pruebas de DAS-ELISA
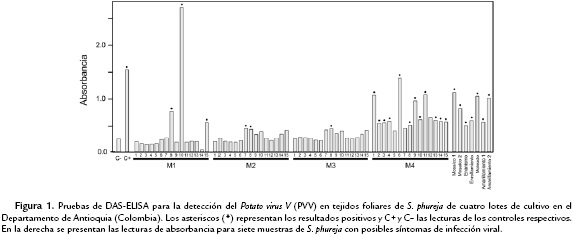
En este trabajo se evaluó la presencia del PVV en muestras foliares de S. phureja obtenidas de cuatro lotes de cultivo ubicados en tres municipios del departamento de Antioquia. Ya que en plantas de papa es frecuente la ocurrencia de infecciones virales mixtas (Wang et al., 2011), es difícil asociar la infección de un virus particular con síntomas genéricos como mosaicos, moteados, amarillamientos o enanismos, razón por la que visualmente no fue posible identificar una sintomatología específica con la ocurrencia de PVV en las muestras bajo análisis. Sin embargo, los resultados de las pruebas de DAS-ELISA, indicaron inequívocamente la infección de PVV en muestras de tejido foliar en los cuatro lotes evaluados, aunque con diferencias evidentes entre los niveles de incidencia (lote M1: 26,7 %, lote M2: 13,3 %, lote M3: 6,7 %, lote M4: 86 %) (Fig. 1). La validez de las pruebas fue confirmada por los altos valores de absorbancia obtenidos para los controles positivos (>1,5) y los bajos niveles alcanzados para los controles negativos (promedio=0,26; SD=0,05); siendo definido con la metodología de Bioreba, un valor de 0,45 como punto de corte para la definición de las pruebas positivas. Adicionalmente, el PVV fue encontrado en las muestras representando las siete muestras sintomáticas, con valores de absorbancia superiores a 0,52 (Fig. 1).
Secuenciación NGS
La secuenciación NGS del transcriptoma de S. phureja generó una librería pareada de 9101996 reads y un total de 1820399200 pb. La librería se depuró por calidad (Phred ≥30) con el software SeqTK y mediante BlastN se seleccionaron los reads asociados a genomas de virus de ARN; de esta forma, 1147244 reads alinearon de manera significativa (e-value < 1E-10) con el genoma de PVV. Para el ensamblaje del genoma se tomó un subconjunto aleatorio de 100000 reads pareados para ser mapeados con respecto el genoma de referencia de PVV-Phureja, obteniéndose una profundidad promedio de 126X (Fig. 2). Durante el análisis de anotación del genoma viral, fue evidente de los patrones de polimorfismo encontrados para cada posición del genoma, la presencia de dos variantes de PVV en los tejidos de papa, con proporciones de (~72 % (Var_A) y ~28 % (Var_B); sin embargo éstas presentan altos niveles de identidad (99,7 % y 31 sustituciones). El genoma de ambas variantes tiene 9828 nt (excluyendo la cola poli-A), con un marco de lectura abierto (ORF) de 9198 nt que codifica para una poliproteína de 3066 aminoácidos y secuencias de 185 y 445 nt en sus extremos UTR 5'y 3', respectivamente.
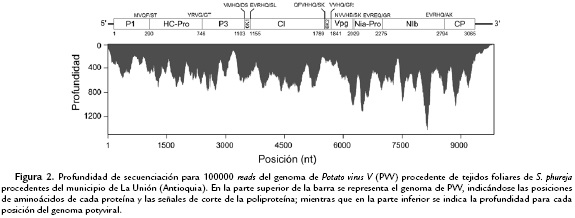
Las predicciones de las secuencias de las proteínas P1, proteinasa HC-Pro, P3, PIPO, 6K1, proteína de inclusión cilíndrica (CI), 6K2, proteína de unión al genoma (VPg), proteinasa a de inclusión nuclear (NIa-Pro), replicasa viral (NIb) y proteína de la cápside (CP) tienen 289, 456, 357, 247, 52, 634, 52, 188, 246, 519 y 272 aa, respectivamente y los sitios de corte proteolíticos se identificaron en los residuos 289, 746, 1130, 1155, 1789, 1841, 2029, 2275 y 2794 (Fig. 2).
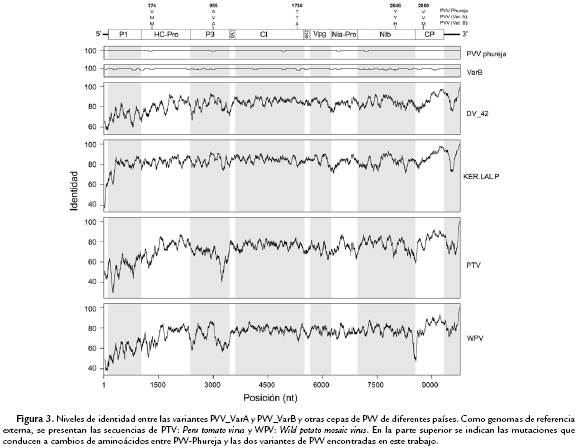
La variante A de PVV presentó cinco mutaciones puntales respecto al genoma de la cepa PVV-Phureja, aunque éstas sólo representaron dos cambios de aminoácidos a nivel de la poliproteína: V374M (HC-Pro) y A955V (P3). En el genoma de la variante B por su parte, se identificaron 28 mutaciones puntuales con respecto a la cepa PVV-Phureja y tres cambios de aminoácidos en la poliproteína: T1736A (CI), Y2646H (NIb), V2860M (CP) (Fig. 3).
Los motivos y regiones que caracterizan el genoma de los potyvirus fueron identificados en ambas variantes de PVV, e incluyeron los motivos conservados en HC-Pro FRNK (residuos de aa 468-471) e IGN (residuos 537-539 aa) asociados a la supresión de silenciamiento de genes y replicación viral, respectivamente; la secuencia nucleotídica GA7C en la posición (2908-2918) de P3, resultante de un cambio en el marco de lectura que da origen a la proteína P3N-PIPO (232 aa); los motivos GSGKSTGLP (residuos 1243-1250) y DECH (residuos 1328-1331) en CI, que indican la homología de esta proteína con la superfamilia de helicasas SF2; los sitios de corte de la cisteín proteasa NIa-Pro en las uniones P3/6K1, 6K1/CI, CI/6K2, 6K2/VPg, VPg/NIa-Pro, NIa-Pro/NIb, NIb/CP con secuencia de reconocimiento V-X-(HE)-(QE); el motivo GDD conservado en las RdRp virales (residuos 2625-2627) y las secuencias de localización nuclear en NIb: NSLI consiste de los residuos 1-16 con la secuencia GRSTGWMLNALKENLQ, y la NSLII de TPISTPDGTIIKKFRGNNSGQPSTV (residuos 2566-2590). Finalmente el motivo DAG (residuos 2802-2805) en la región CP, que está asociado a la transmisión por áfidos.
Los análisis de variación realizados entre la secuencias de las dos variantes de PVV encontradas en este trabajo y las tres cepas con genomas completos reportados para PVV en el mundo (DV42, KER.LAL.P y PVV-Phureja) indican niveles de identidad a nivel de nucleótidos del 82-83 % con las dos primeras, siendo las regiones que codifican para P1 y para las proteinasas Hc-Pro y NIa-Pro las más disímiles, con niveles de 71 a 83 %; mientras que para las comparaciones realizadas con respecto a la cepa PVV-Phureja, dichos niveles fueron superiores al 99 % para todos los genes (Fig. 3). Para este último caso, fue además notable el hecho que ocho de las diez proteínas putativas de dichas cepas compartieron el 100 % de identidad a nivel de aminoácidos, y los dos restantes (HC-Pro y P3) del 99,7 %.
Detección de PVV por RT-qPCR
Con el uso de los cebadores diseñados para la detección de PVV mediante RT-qPCR se determinó la infección de PVV en seis de las muestras evaluadas, incluyendo el control positivo, dos de las muestras sintomáticas (amarillamiento y mosaico) y en tres de aquellas asintomáticas. Los valores de Ct oscilaron entre 12,08 y 21,86, mientras que los de Tm se presentaron en el rango de 78,7 °C a 80,2 °C (Tabla 1). La naturaleza de cuatro de dichos amplicones (incluyendo el control positivo) fue confirmada por secuenciación Sanger. Se encontró por comparación con las bases de datos moleculares, que dichas secuencias correspondían a una región de cerca de 50 pb de la cápside viral de diferentes aislamientos de PVV (Ej. Accesiones gb: KC438301, KC438303 y KC438304), con niveles de identidad de 92 a 96 % y valores de e en el rango de 9x10-4 a 5x10-5.
Detección de PVV por RT-PCR
Los cebadores diseñados (PVV-phu-F y PVV-phu-R) para la detección de PVV mediante RT-PCR convencional, fueron igualmente validados en el trabajo, al lograrse la amplificación de fragmentos del tamaño esperado (459 pb) en cinco de los grupos de muestras evaluadas y en el control positivo (Tabla 1). Las secuencias obtenidas para cuatro de los amplicones, confirmaron la identidad de dichas regiones como parte del gen CP de PVV, con niveles de identidad del 82-83 % (valores e=1x10-115 a 2x10-118, cobertura= 99 %) con respecto a diversos aislamientos de este virus cuyas secuencias se encuentran depositadas en las bases de datos moleculares (Ej. Accesiones gb: KC438301, KC438303, GU144345).
Análisis filogenéticos
Los análisis filogenéticos generados con las dos metodologías evaluadas en el estudio (NJ y ML) para las cinco secuencias de CP obtenidas a partir de RT-PCR convencional y NGS, generaron dendrogramas con topologías similares, por lo que sólo se presenta la descripción para el que utilizó NJ (Fig. 4). El dendrograma presentó una topología que separó los aislamientos de PVV (90 % bootstrap) de aquellos representando diferentes potyvirus utilizados como grupos externos de análisis. El clado de PVV se dividió a su vez en dos subgrupos principales y una rama independiente; subdivisiones éstas fuertemente soportadas por valores de bootstrap superiores a 99 %. El primer subclado presentó dos ramas principales, una mayoritaria que incluyó 12 cepas de PVV de Eurasia (Ej. Escocia, Holanda, Irán, etc.) y la otra con un sólo aislamiento del Perú (AJ516022). El segundo subclado incluyó todos los aislamientos de PVV obtenidos en la presente investigación sobre S. phureja en Colombia, con 100 % de soporte de bootstrap, así como el aislamiento PVV-Phureja. La rama independiente, se presentó entre ambos subclados y correspondió a un único aislamiento PA-4 procedente de la variedad Papa Amarillo y reportado en un análisis cuarentenario en EEUU (AY521595) (Shiel et al., 2004). Por otra parte, el análisis realizado con respecto a los grupos externos de referencia, indicó que PVV se presenta como un virus filogenéticamente más relacionado con ERV y PTV que con WPMV, BsMoV o PepYMV (Fig. 4). Las matrices de identidad obtenidas partir de estas secuencias de CP, confirmaron los resultados del análisis filogenético, al encontrarse que para las cepas de PVV obtenidas en S. phureja en Colombia, dichas secuencias fueron prácticamente idénticas (>99,7 %), mientras que tan sólo compartieron entre 82,8 y 84,7 % de identidad con respecto a cepas de PVV de otras procedencias geográficas y valores inferiores a 78,8 % con relación a la secuencias de otros potyvirus incluidos como referencias externas del trabajo (Fig. 4).
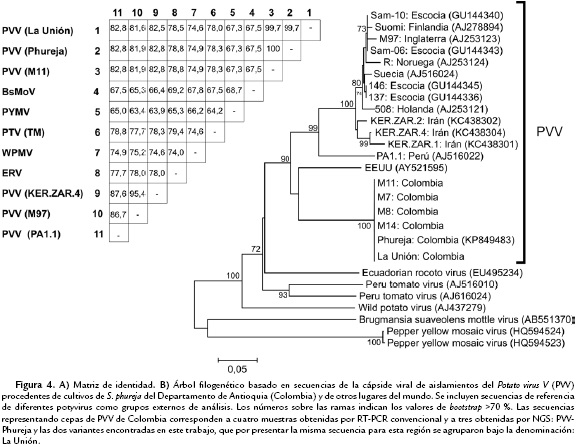
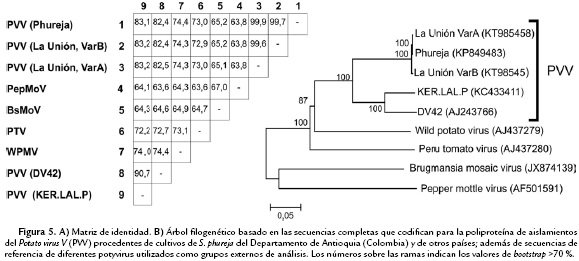
El análisis filogenético realizado con base en la secuencia completa del ORF que codifica para la poliproteína de PVV y otros potyvirus, presentó una topología similar a la encontrada para CP, al separar en dos subclados (100 % bootstrap) a la especie PVV; uno de ellos ocupado por las cepas procedentes de Eurasia y el otro por aquellas obtenidas en Colombia sobre S. phureja. Así mismo, el análisis con respecto a otros potyvirus de referencia, confirmó la validez taxonómica de PVV como una especie claramente diferenciada en el género Potyvirus (Fig. 5). Con base en estos análisis filogenéticos de secuencias de CP y de la poliproteína completa, sugerimos denominar el nuevo subclado identificado en este trabajo como el linaje filogenético Phureja de PVV (PVVPhu).
DISCUSIÓN
A pesar de que PVV es uno de los virus que se reportan con altos niveles de incidencia en variedades locales de papa en países como Irán (32,9 % en S. tuberosum cv. Zardi) (Shamsadden-Saeed et al., 2014) y Costa Rica (37 % en S. tuberosum) (Vásquez et al., 2006), y a que ha sido declarado como virus cuarentenario para China, el principal productor de papa del mundo con cerca del 26,3% del área global destinada a este cultivo (Wang et al., 2011), son muy pocos los estudios en los que se han evaluado sus características genómicas y sus niveles de variación intraespecíficos. Esta situación se ve claramente reflejada por el reporte de tan sólo tres genomas completos de PVV en el mundo: DV42 de Escocia (Oruetxebarria et al., 2000), KER.LAL.P de Irán (Shamsadden-Saeed et al., 2014), y PVV-Phureja de Colombia (Gutiérrez et al., 2016). En este último estudio, se secuenció el transcriptoma de tejido foliar de una sola planta de S. phureja var. Criolla Colombia de La Unión (Antioquia) que presentaba síntomas de mosaicos suaves y se encontró que el genoma de PVV presentaba niveles de identidad del 83 % con respecto a los otros dos aislamientos secuenciados completamente; además, se hallaron diferencias de hasta el 5 % en la secuencia de aa de CP entre dicha cepa y otras de diferentes orígenes geográficos. Estos resultados, evidentemente señalaban la ocurrencia de niveles de variación no reportados previamente para esta especie viral y por tanto en el presente estudio se le dio continuidad a dicho reporte, pero en este caso secuenciando el transcriptoma de un grupo de muestras foliares de diferentes plantas de S. phureja, procedentes del mismo municipio de La Unión. Esta estrategia resultó exitosa, por cuanto se encontraron dos variantes principales de PVV (Var_A y Var_B) en dicha muestra compuesta; las que a pesar de presentar altos niveles de identidad (99,7 %), tuvieron 31 nucleótidos diferentes a lo largo de sus genomas de 9828 nt. La secuencia de la variante mayoritaria (Var_A, 78 %) se presentó como más cercana a la cepa PVV-Phureja, al sólo presentar cinco mutaciones puntuales con respecto a ésta, mientras que la variante B tuvo 28 mutaciones, tres de las cuales conducían a cambios putativos en la secuencia de aa de la poliproteína. Los análisis filogenéticos realizados tanto para la poliproteína completa como para el gen CP de PVV, confirmaron la ocurrencia de un subclado en esta especie viral ocupado exclusivamente por cepas de Colombia; dicho subgrupo comparte bajos niveles de identidad (de 82,8 a 84,7 %) con las demás cepas de PVV bajo análisis, incluyendo aquellas de Perú reportadas por Spetz et al., (2003). El hallazgo de este linaje genético de PVV, que se propone denominar en el presente estudio como PVVPhu, había sido inicialmente inferido por Gutiérrez et al. (2014), en el primer reporte formal de este virus en Colombia. En dicho trabajo, utilizando pirosecuenciación 454 GS-FLX del transcriptoma de tejido radicular de S. phureja, se detectó la presencia de PVV en el 6,5 % de los reads de CP allí obtenidos. Así mismo, con base en los análisis filogenéticos de dichas secuencias, se estableció la ubicación conjunta en un subclado de esta variante Colombiana de PVV con la cepa PA-4 reportada por Shiel et al., (2004) a partir de un material del cultivar Papa amarilla de S. tuberosum que presentaba síntomas de mosaico en una estación cuarentenaria de EEUU.
Los estudios moleculares realizados recientemente con PVV en Colombia (Gutiérrez et al., 2014; Gutiérrez et al., 2016, este estudio), y en Irán (Shamsadden-Saeed et al., 2014), donde se encontró un grupo de cepas infectando la variedad local Agria de S. tuberosum con <97,5 % de identidad en la secuencia de CP con respecto a los aislamientos reportados de este virus, amplían el panorama general sobre los niveles de variación intraespecífica de PVV en el mundo; que con excepción de los reportes de Spetz et al., (2003) en Perú, consideraban al genoma de este virus como altamente conservado (Oruetxebarria et al., 2000; Oruetxebarria et al., 2001; Mortensen et al., 2010). En dichos análisis filogenéticos se soporta la validez taxonómica de esta especie dentro del género Potyvirus (Inoue-Nagata et al., 2002; Spetz et al., 2003), situación que también fue evidente en el presente trabajo, donde todas las cepas de PVV conformaron un clado monofilético, siendo el ERV y PTV las especies de potyvirus más cercanas filogenéticamente.
Esta información genómica no sólo resulta útil para resolver dudas sobre aspectos filogenéticos y taxonómicos en los virus, sino también que sirve de base para ajustar los métodos de diagnóstico. Así por ejemplo, el hallazgo de la antes referida variante PA-4 de EEUU, se derivó de un falso negativo luego de utilizar anticuerpos comerciales para PVV que no lograron detectar este virus en muestras sintomáticas de la variedad Papa amarilla, lo que posteriormente fue encontrado se debía a la ocurrencia de varios cambios en la secuencia de aa del extremo N-terminal de CP (Shiel et al., 2004), la región más antigénica de los potyvirus (Shukla et al., 1988). En forma similar, la identificación del linaje PVVPhu que se realizó en el presente trabajo, nos condujo al diseño de cebadores para lograr su detección específica, tanto en pruebas de RT-qPCR (PVV_phu_F y qPVV_phu_R ) como de RT-PCR convencional (PVV-phu-F y PVV-phu-R), confirmándose su utilidad práctica tanto a partir de muestras sintomáticas como asintomáticas de S. phureja. Estas pruebas moleculares se constituyen en un aporte para el apoyo a los programas de certificación de tubérculos-semilla en Colombia y otros países cultivadores de papa y una herramienta de utilidad para adelantar estudios de epidemiología y resistencia varietal a PVV en los agroecosistemas andinos. De esta forma, evaluaciones del rango de hospedantes, métodos de transmisión y niveles de incidencia de PVV en cultivos de S. phureja, resultarán fundamentales para establecer un programa de manejo integrado de esta enfermedad viral. Sobre este último aspecto, en el presente estudio utilizando pruebas de DAS-ELISA de la compañía Agdia, se detectó la infección de PVV en los cuatro lotes evaluados, aunque con un alto nivel de variación entre dichos cultivos (de 6,7 % a 86 %). Ya que esta es sólo una primera aproximación a la incidencia que presenta PVV en el país, de gran interés resultará en el futuro próximo, la ejecución de un estudio de incidencia que incluya un alto número de lotes de cultivo de las principales zonas productoras de papa (S. phureja y S. tuberosum) no sólo de Antioquia, sino de otros departamentos como Cundinamarca, Boyacá y Nariño, y que utilice además de pruebas serológicas, las técnicas moleculares aquí descritas; de manera que se ofrezca un mejor panorama sobre la distribución y los niveles de infección de este virus en los cultivos de papa del país; siendo fundamental evaluar sí la variante PVVPhu es exclusiva de S. phureja, o sí igualmente infecta variedades locales de S. tuberosum como Diacol-Capira, ICA-Puracé y Parda-Pastusa.
CONCLUSIONES
En este trabajo se secuenció completamente el genoma de dos variantes de PVV obtenidas de un grupo de muestras foliares de S. phureja con síntomas de mosaicos suaves, utilizando el sistema Illumina HiSeq 2000. Este genoma de 9828 nt codifica para un poliproteína de 3066 aa y presenta bajos niveles de identidad (82-83 %) con respecto a las dos secuencias completas disponibles en el mundo para este virus; así como alta homogeneidad con la cepa PVV-Phureja de Colombia, por lo que se propone denominar a este subgrupo de cepas como el linaje genético PVVPhu.
Con base en los genomas secuenciados de PVVPhu, se diseñaron dos pares de cebadores útiles para su detección específica en pruebas de RT-qPCR y RT-PCR convencional. Estas pruebas ofrecen alternativas para el diagnóstico de este virus en laboratorios que disponen de termociclador en tiempo real y en aquellos que sólo cuentan con los equipos básicos de PCR; y además permitirán ampliar los estudios filogenéticos y de caracterización molecular de PVV en diferentes regiones productoras de papa de Colombia y de otros países.
AGRADECIMIENTOS
Este trabajo fue financiado por la Vicerrectoría de Investigaciones de la Universidad Nacional de Colombia (Proyecto 28616) y por International Foundation for Science (IFS) de Suecia (Proyecto C/4634-2).
REFERENCIAS
BIOREBA. ELISA data analysis. Available in: http://www.bioreba.com. Cited: 30th May 2015. [ Links ]
Calvert ELP, Cooper P, McClure J. An aphid transmitted strain of PVYc recorded in potatoes in Northern Ireland. Rec Agric Res. 1980;28:63-74. [ Links ]
Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783-791. Doi:10.2307/2408678. [ Links ]
Fribourg CE, Nakashima J. Characterization of a new Potyvirus from potato. Phytopathology. 1984;74(11):1363-1369. Doi:10.1094/Phyto-74-1363. [ Links ]
García D, Gutiérrez P, Marín M. Análisis filogenético y variabilidad molecular del Potato virus X (PVX) en cultivos de papa de Antioquia. Acta biol. Colomb. 2016;21(1):111-122. Doi:10.15446/abc.v21n1.51398. [ Links ]
Gil JF, Cotes JM, Marín M. Detección serológica y caracterización molecular de Potato virus S (PVS, Carlavirus) en cultivos de papa de Colombia. Rev Biol Trop. 2013;61(2):565-575. [ Links ]
Gish W, States DJ. Identification of protein coding regions by database similarity search. Nature Genet. 1993;3:266-272. Doi:10.1038/ng0393-266. [ Links ]
GitHub. SeqTK. Available in: https://github.com/lh3/seqtk/. Cited: 15 Feb 2015. [ Links ]
Gutiérrez PA, Alzate JF, Marín M. Complete genome sequence of a novel Potato virus S strain infecting Solanum phureja in Colombia. Arch Virol. 2013;158(10):2205-2208. Doi:10.1007/s00705-013-1730-7. [ Links ]
Gutiérrez PA, Alzate JF, Marín M. Caracterización del viroma de ARN de tejido radical de Solanum phureja mediante pirosecuenciación 454 GS-FLX. Bioagro; 2014;26(2):89-98. [ Links ]
Gutiérrez-Sánchez P, Jaramillo-Mesa H, Marín-Montoya M. Genome sequence of a divergent Colombian isolate of potato virus V (PVV) infecting Solanum phureja. Acta Virol. 2016;60(1):49-54. Doi:10.4149/av_2016_01_49. [ Links ]
Guzmán-Barney M, Franco-Lara L, Rodríguez D, Vargas L, Fierro JE. Yield losses in Solanum tuberosum Group Phureja cultivar Criolla Colombia in plants with symptoms of PYVV in field trials. Am J Pot Res. 2012;89(6):438-447. Doi:10.1007/s12230-012-9265-0. [ Links ]
Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999;41:95-98. [ Links ]
Hernández-Guzmán AK, Guzmán- Barney M. Detección del virus del amarillamiento de las nervaduras de la hoja de la papa en diferentes órganos de Solanum tuberosum grupo Phureja cv Criolla Colombia utilizando RT-PCR convencional y en tiempo real. Rev Colomb Biotecnol. 2014;16(1):74-85. Doi:10.15446/rev.colomb.biote.v16n1.44226. [ Links ]
Inoue-Nagata AK, Fonseca MEN, Resende RO, Boiteux LS, Monte DC, Dusi AN, De Avila AC, Van der Vlugt RAA. Pepper yellow mosaic virus, a new potyvirus in sweetpepper, Capsicum annuum. Arch.Virol. 2002;147(4):849-855. Doi:10.1007/s007050200032. [ Links ]
Jones RAC, Fuller NJ. Incidence of Potato virus V in potato stocks in England and Wales. Plant Pathol. 1984;33(4):595-597. Doi:10.1111/j.1365-3059.1984.tb02885.x. [ Links ]
Kreuze J, Koenig R, De Souza J, Vetten HJ, Muller G, Flores B, Ziebell H, Cuellar W. The complete genome sequences of a Peruvian and a Colombian isolate of Andean potato latent virus and partial sequences of further isolates suggest the existence of two distinct potato-infecting tymovirus species. Virus Res. 2013;173(2):431-435. Doi:10.1016/j.virusres.2013.01.014. [ Links ]
Langmead B, Salzberg S. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357-359. Doi:10.1038/nmeth.1923. [ Links ]
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H. Clustal W and Clustal X version 2.0. Bioinformatics. 2007:23:2947-2948. [ Links ]
Medina HC, Gutiérrez PA, Marín M. Detección del Potato virus Y (PVY) en tubérculos de papa mediante TAS-ELISA y qRT-PCR en Antioquia (Colombia). Bioagro. 2015;27:83-92. [ Links ]
Mortensen RJ, Shen X, Reid A, Mulholland V. Characterization of viruses infecting potato plants from a single location in Shetland, an isolated Scottish archipelago. J Phytopathol. 2010;158(9):633-640. Doi:10.1111/j.1439-0434.2010.01672.x. [ Links ]
Oruetxebarria I, Kekarainen T, Spetz C, Valkonen JPT. Molecular characterization of Potato virus V genomes from Europe indicates limited spatiotemporal strain differentiation. Phytopathology. 2000;90(4):437-444. Doi:10.1094/PHYTO.2000.90.4.437. [ Links ]
Oruetxebarria I, Valkonen JP. Analysis of the P1 gene sequences and the 3-terminal sequences and secondary structures of the single-stranded RNA genome of Potato virus V. Virus Genes. 2001;22(3):335-343. Doi:10.1023/A:1011174509453. [ Links ]
Rodríguez LE, Ñustez CE, Estrada N. Criolla Latina, Criolla Paisa y Criolla Colombia, nuevos cultivares de papa criolla para el departamento de Antioquia (Colombia). Agron Col. 2009;27(3):289-303. [ Links ]
Rodríguez OJ, Rodríguez LE, Cotes JM. Caracterización morfo agronómica de híbridos provenientes del cruzamiento entre especies diploides de papa. Rev Fac Ciencias Básicas. 2013;9(1):54-67. [ Links ]
Shamsadden-Saeed F, Massumi H, Moradi S, Maddahian M, Heydarnejad J, Pour AH, Varsani A. Incidence and characterization of Potato virus V infections in Iran. Virus Dis. 2014;25(1):78-84. Doi:10.1007/s13337-013-0178-4. [ Links ]
Shiel PJ, Miller L, Slack SA, Berger PH. Isolation and partial nucleic acid characterization of a new isolate of Potato virus V with distinct biological and serological properties. Plant Dis. 2004;88(4):368-372. Doi:10.1094/PDIS.2004.88.4.368. [ Links ]
Shukla DD, Strike PM, Tracy SL, Gough KH, Ward CW. The N and C termini of the coat proteins of potyviruses are surface-located and the N terminus contains the major virus-specific epitopes. J Gen Virol. 1988;69(7):1497-1508. Doi:10.1099/0022-1317-69-7-1497. [ Links ]
Spetz C, Taboada AM, Darwich S, Ramsell J, Salazar LF, Valkonen JP. Molecular resolution of a complex of potyviruses infecting solanaceous crops at the centre of origin in Peru. J Gen Virol. 2003;84(9):2565-2578. Doi:10.1099/vir.0.19208-0. [ Links ]
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular evolutionary genetics analysis. Mol Biol Evol. 2013;30(12):2725-2729. Doi:10.1093/molbev/mst197. [ Links ]
Vásquez V, Montero-Astúa M, Rivera C. Incidencia y distribución altitudinal de 13 virus en cultivos de Solanum tuberosum (Solanaceae) en Costa Rica. Rev Biol Trop. 2006;54:1135-1141. [ Links ]
Wang B, Ma Y, Zhang Z, Wu Z, Wu Y, Wang Q, Li, M. Potato viruses in China. Crop Prot. 2011:30(9):1117-1123. Doi:10.1016/j.cropro.2011.04.001. [ Links ]
Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden T. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 2012:13:134. Doi:10.1186/1471-2105-13-134. [ Links ]

