











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
INTRODUCCIÓN
Plasticidad celular
Tradicionalmente se ha pensado que los organismos multicelulares se caracterizaban por un destino predeterminado y fijado de diferenciación celular (Bloom, 1937). Incluso los estudios de diferenciación celular se convirtieron en el centro de atención de toda una sociedad que fue creada en 1971 (International Society of Differentiation, 2019). Luego se creó una revista dedicada a tal misión en 1973, pues un campo tan diverso y amplio como la diferenciación celular requería de aproximaciones multidisciplinares, porque la generalidad de las teorías no estaba aportando guías experimentales para el futuro de la investigación (Stewart, 1973).
Aunque la identidad celular es una característica distintiva de todas las células, históricamente existen evidencias de que, ante una lesión, muchos organismos de distintos phyla pueden convertir un tipo celular en otro, lo que se conoce como plasticidad celular (Tata y Rajagopal, 2016). Comentan los autores del anterior trabajo, que desde 1712 Abraham Trembley estudió cómo las hidras regeneraban partes enteras de su organismo y en el proceso había una transición dada por la plasticidad celular, como fue extensamente ilustrado en su libro "Mémoires pour servir à l'histoire d'un genre de polypes d'eau douce, à bras en forme de cornes" disponible en internet (Trembley, 1744).
La diferenciación celular fue estudiada durante más de dos siglos por embriólogos y otros investigadores, pero curiosamente una luz que fue profética para los descubrimientos actuales la constituyó el texto de Konrad Waddington (1957). Antes del descubrimiento de la estructura del DNA Waddington conjeturó, de manera elegante y precisa mediante modelos matemáticos, que la diferenciación celular ocurría como un fenómeno probabilístico, en un paisaje epigenético en el que los destinos celulares eran representados por valles entre picos adaptativos. Los genes en su estrategia (como se titulaba su libro), explicaban una parte del fenómeno, pero el destino celular diferenciado subyacía en fenómenos dinámicos y probabilísticos.
Durante el siglo XX varios hechos confluyeron para que la diferenciación celular dejara de ser un proceso irreversible y, como hipotetizan Kraft y Rubin (2016), la concesión del premio Nobel de Medicina de 2012 por la investigación en células madre dejó explícito que la plasticidad celular era el fenómeno que subyacía a estos importantes descubrimientos. El término plasticidad celular se usó por primera vez en 1985 por Helen Blau et al. (1985); ella fue pionera en tales investigaciones, aunque hizo parte durante muchos años del comité de la Sociedad Internacional para la Diferenciación celular (National Academy of Sciences, 2016).
Entre las diversas posibilidades de plasticidad celular, la más interesante es el fenómeno de dediferenciación mediante el cual un linaje establecido puede regresar en su fenotipo diferenciado hacia uno más indiferenciado (Tata y Rajagopal, 2016). Una de tales alternativas la constituye la Transición Epitelio-Mesénquima, o más conocida por sus siglas en inglés como EMT.
La EMT es un proceso altamente conservado en vertebrados, y ambos tejidos, epitelio y mesénquima, se encuentran en casi todos los órganos de dicho grupo taxonómico (Hay, 2005). Consiste en que las células epiteliales cambian su fenotipo, hacia un comportamiento migratorio e invasivo, más característico de células mesenquimatosas (Nieto, 2011). Recientemente, se le considera un programa celular importante, tanto en los procesos normales como en la embriogénesis, la organogénesis y el sellamiento de heridas, como en la progresión tumoral (Barriere et al., 2015). Además, la EMT está involucrada en la inducción de metástasis (Talbot et al., 2012; Barriere et al., 2015) y representa una vía alterna al desarrollo de eventos oncogénicos tempranos.
Hasta ahora se han descrito tres tipos de EMT dependiendo del contexto fisiológico: EMT tipo I, relacionada con embriogénesis y desarrollo de órganos (Barriere et al., 2015); EMT tipo II, asociada a regeneración o reparación de tejidos y fibrosis; y EMT tipo III, concerniente a la progresión de cáncer y propiedades de células madre cancerígenas (Qi et al., 2016). Tanto la EMT como las variantes mencionadas se llevan a cabo con la participación de los mismos genes y vías de señalización (Welch-Reardon et al., 2015), que actúan de modo similar en el tiempo y espacio celular y además constituyen fenómenos conservados evolutivamente. Tal vez por ello, Weinberg considera a la EMT como un "programa celular" (Dongre y Weinberg, 2019).
Tradicionalmente, los virólogos han descrito el efecto citopático como un cortejo de cambios morfológicos producidos por los virus que infectan células animales y que son visibles al microscopio óptico (Luria et al., 1953). La formación de sincitios -fusión de membranas celulares entre varias células- por ejemplo, es un evento característico de virus con envolturas, y sobre todo de los virus RNA, que usan el sistema de endomembranas para hacer sus complejos replicativos (Den Boon y Ahlquist, 2010).
Sin embargo, durante varios decenios no se prestó mucha atención a las células hospederas, mientras ocurría el mencionado efecto citopático. Empezando el milenio nacieron nuevos eventos científicos y revistas como "Cellular Microbiology", que claramente estaban mostrando un interés renovado por lo que les sucede a las células bajo un evento de infección viral.
En ese orden de ideas, al ser invasores intracelulares los virus subvierten numerosos mecanismos de la fisiología, por lo cual no resulta extraño que sean inductores de la EMT. Podría haber sido uno de los hallazgos tradicionales del efecto citopático, que no estaba categorizado como EMT, sino como "alargamiento celular, o elongación celular". Se tienen diversos miembros que han sido descritos como inductores de la EMT, los cuáles se mencionarán más adelante.
A continuación, profundizaremos en los detalles más importantes para el entendimiento de esta relación virus-hospedero. Para ello, se realizó una búsqueda y selección sistemática de la información en bases de datos especializadas, en las que se rastrearon las publicaciones base, las más claves y relevantes, además de lo último reportado por los expertos de cada subtema. Este artículo de revisión recorre el proceso de EMT inducida por virus, desde lo fenotípico a lo molecular, para finalmente aclarar el panorama actual como base para nuevas estrategias de prevención y control de enfermedades infecciosas.
TEJIDO EPITELIAL Y MESENQUIMATOSO
Algunas consideraciones evolutivas, estructurales y funcionales
Los deuterostomados, miembros de los cordados, están compuestos de tres tipos de tejidos (ectodermo, mesodermo y endodermo). Sin embargo, la aparición de células mesenquimatosas con la habilidad de invadir la matriz extracelular (EM) podría haber sido la novedad evolutiva que permitió producir formas corporales altamente complejas en el subphylum Vertebrata (Hay, 1995). Es importante recordar que las células mesenquimales derivadas del mesodermo son necesarias para generar los endoesqueletos (cartílago y hueso) en los vertebrados, que ofrecen un patrón interno más intrincado y mayores tamaños corporales, que los propiciados por exoesqueletos como en artrópodos (Hay, 1995).
El tejido epitelial está caracterizado por células estrechamente unidas a sus vecinas y poca matriz extracelular entre ellas (Kardong, 2011). Aunque existen diversos tipos de tejido epitelial (Junqueira y Carneiro, 2015), aquí nos centraremos en un tipo especial que está construido de células polarizadas, como las del epitelio bronquial o el intestinal. Esta clase de epitelio posee una arquitectura supracelular organizada (Hay, 2005), ya que todos sus orgánulos se encuentran dispuestos siguiendo un fenotipo polarizado en sentido apico-basal (Hay, 1995). Este fenotipo epitelial no solo responde a la disposición de orgánulos, sino también a la de proteínas de adherencia distribuidas en la membrana plasmática; en la parte basal, las células están unidas a la matriz extracelular mediante integrinas asociadas a filamentos intermedios; baso-lateral y lateralmente, poseen uniones adherentes (E-cadherina), además de uniones estrechas y desmosomas; y en la parte apical se caracterizan por poseer prolongaciones de filamentos de actina (Rodriguez-Boulan y Nelson, 1989). Además de ser una característica estructural, la polarización está configurada funcionalmente porque el transporte de proteínas sucede de una manera vectorial, en sentido basal hacia el apical (Gumbiner, 1990). No obstante, se ha descubierto que la versatilidad de las células epiteliales polarizadas en su tráfico de proteínas es mucho mayor (Rodriguez-Boulan et al., 2005). Las células polarizadas así dispuestas eventualmente pueden migrar mediante un proceso coordinado, conocido como migración colectiva (Wong et al., 2014; Mayor y Etienne, 2016), regulada principalmente por E-cadherina (Shih y Yamada, 2012).
Por el contrario, el tejido mesenquimatoso posee baja expresión de las proteínas de unión y de citoesqueleto; este tejido se caracteriza por expresar N-cadherina, filamentos intermedios de vimentina y otros marcadores mesenquimatosos. Las células mesenquimatosas poseen una polaridad antero-posterior, caracterizada por un borde que avanza o lamelipodio, que lidera el proceso de migración individual (Lamouille et al., 2014), lo cual les permite a las células embebidas en la matriz extracelular migrar rápidamente (Talbot et al., 2012; Qi et al., 2016).
TRANSICIÓN EPITELIO-MESÉNQUIMA (EMT)
La EMT se inicia con la represión de la expresión de las proteínas de unión célula-célula y célula-matriz extracelular (principalmente E-cadherina). Le sigue la reorganización del citoesqueleto de actina, la expresión de marcadores mesenquimatosos, como las proteínas de adhesión, N-cadherina y β-catenina, y la sobreexpresión de filamentos intermedios de vimentina. Finalmente, las células adquieren la polaridad antero-posterior de células mesenquimatosas caracterizada por la formación de lamelipodios y la secreción de metaloproteinasas (MMP). Estas últimas se encargan de degradar la matriz extracelular de la membrana basal en la cual se encuentra embebido el tejido (Lamouille et al., 2014) (Figs. 1 y 2a-b), lo que favorece la motilidad (Gonzalez y Medici, 2014; Qi et al., 2016) y la resistencia a la apoptosis (Qi et al., 2016), característica de los carcinomas malignos, los cuales son los tumores más invasivos (Nieto, 2017).
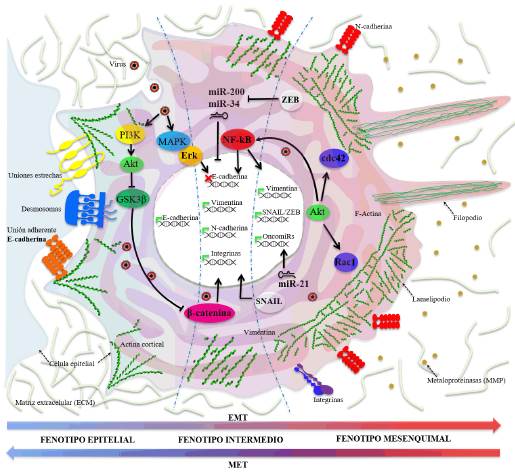
Figura 1 Transición Epitelio-Mesénquima (EMT) y mecanismos moleculares de activación en un evento de infección viral. La punta de la flecha indica activación y la punta en barra indica represión. La equis roja indica represión de la transcripción y las flechas verdes indican activación de la transcripción.
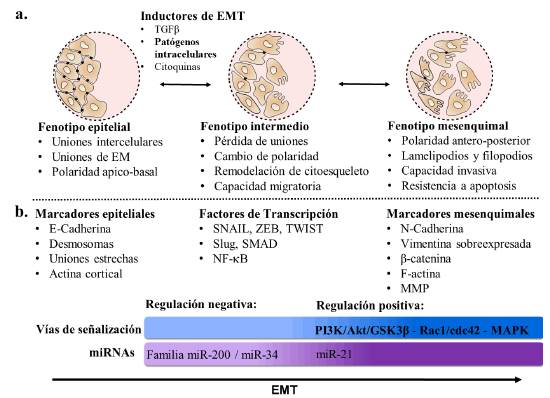
Figura 2 a. Transición fenotípica y estructural de la EMT. b. Actores moleculares más relevantes y su rol en la EMT. Compilación de los eventos fenotípicos y estructurales más característicos durante la EMT, identificación de los marcadores moleculares de cada fenotipo, factores de transcripción más relevantes y rol de los actores moleculares encargados de regular el proceso, positiva o negativamente.
Se ha descrito también el proceso contrario, llamado Transición Mesénquima-Epitelio (MET, Lamouille et al., 2014) (Fig. 1), involucrado en la EMT tipo I, ya que durante el desarrollo embrionario ocurren rondas consecutivas de EMT y MET. El proceso de la MET, por ejemplo, es indispensable para que las células tumorales vuelvan al fenotipo epitelial y favorezcan la colonización de tejidos distantes y la promoción de la metástasis (Nieto, 2011). Este proceso de transición demuestra ser tan versátil que, adicionalmente, puede desarrollarse también en células endoteliales, proceso conocido como Transición Endotelio-Mesénquima (EnMT), en el cual las células conservan uniones intercelulares y migran como una "procesión" de células (Welch-Reardon et al., 2015).
Mecanismos moleculares inductores de la EMT
La EMT puede ser activada por distintos estímulos, pero en general es el resultado de la señalización inducida por factores de crecimiento. Entre sus principales inductores, el más reportado es el TGFβ (Transforming Growth Factor beta), el cual activa rutas como las quinasas PI3K (Phosphoinositide 3-kinase) y Akt (por timoma espontáneo en una cepa de ratones llamada AKR, también llamada PKB, Protein Kinase B), y MAPK (Mitogen-Activated Protein Kinase). Las MAPK están asociadas al efector Erk (Extracellular signal-Regulated Kinase) (Gonzalez y Medici, 2014), median procesos de proliferación, diferenciación y migración, y promueven la activación de SNAIL/Slug (Talbot et al., 2012; Gonzalez y Medici, 2014). SNAIL es un factor de transcripción, fuerte represor de la transcripción del gen de E-cadherina.
En cuanto a los factores de transcripción más importantes se encuentran: SNAIL, ZEB (Zinc finger E-box-binding homeobox), TWIST y NF-kB (Nuclear Factor kappa-light-chain-enhancer of activated B cells), además de Slug y SMAD. El TGFβ (Moustakas y Heldin, 2014; Wang et al., 2014) media también la activación de SMAD2/3 (Lamouille et al., 2012; Talbot et al., 2012) y el factor de transcripción NF-kB es inductor de marcadores mesenquimatosos (Moustakas y Heldin, 2014). Adicionalmente, la activación de receptores Tirosina/Quinasa (RTK) por parte de factores de crecimiento, puede también inducir las rutas MAPK (Lamouille et al., 2014) y PI3K/Akt y la ruta de Src quinasas/ ABL (virus del sarcoma de Rous), que promueve la motilidad celular (Lamouille et al., 2014; Álvarez-Díaz et al., 2019).
Por otro lado, los estímulos de la matriz extracelular también pueden activar integrinas que ayudan a mediar la vía del PI3K/Akt y la expresión de MMPs (Matrix Metalloproteinases) (Lamouille et al., 2014) lo que promueve el crecimiento, la proliferación y la motilidad. Se ha encontrado también que la inhibición de RhoA (Ras Homology Family), y con ello de las fibras de tensión, simultáneamente activa a dos RhoGTPasas, Cdc42 y Rac1, que generan proyecciones celulares como lamelipodios y filopodios (Cuartas-López et al., 2018) (Fig. 1) y facilitan la migración y la invasión. Otros estímulos como la inflamación crónica mediante acción constante de citoquinas, puede activar la ruta de STAT3 (Signal transducers and activators of transcription), inductora de SNAI1.
Adicionalmente, se conocen otras vías activadas mediante receptores acoplados a proteínas G (GPCR) (Shi et al., 2012) que promueven la progresión de la EMT. Entre ellas se encuentran la señalización activada por Hedgehog (Hh), que actúa como un morfógeno (Talbot et al., 2012) mediado por TFG-β, y el oncogen Ras (Chen et al., 2011) que activa la Wnt/β-catenina (Talbot et al., 2012; Gonzalez y Medici, 2014). Por otro lado, la deslocalización de la β-catenina de las uniones intercelulares permite su translocación al núcleo para la inducción de la EMT, por medio de su interacción directa con el factor de transcripción LEF1 (lymphoid enhancer factor 1) (Lamouille et al., 2014).
Por último, la ruta de Notch (Talbot et al., 2012) también media la activación del NF-kB y promueven la expresión de SNAIL, que reprime los genes de E-cadherina, la sobre-regulación de genes de vimentina y la activación de la transcripción de MMPs (Fig. 1). Además, las condiciones de hipoxia también pueden activar el factor inducible de hipoxia (HIF1β), que está implicada en la progresión de la EMT (Nieto, 2011; Talbot et al., 2012). En las Figs. 1 y 2a-b, se puede observar una visión general y resumida de los principales actores moleculares implicados y su participación en la activación de la EMT.
Por otro lado, con respecto al proceso de la EnMT, entre sus marcadores están la señalización de TGFβ y Notch, ambos reguladores de angiogénesis bien descritos hasta ahora (Hall y Ran, 2010). Otra ruta es la señalización del factor de crecimiento fibroblástico (FGF), la cual se encuentra entre EnMT y angiogénesis (Dorey y Amaya, 2010; Welch-Reardon et al., 2015). Todas las rutas anteriormente mencionadas ocurren en EMT y EndMT y convergen para finalmente afectar la integridad epitelial o endotelial (Hofman y Vouret-Craviari, 2012), lo que atenúa la respuesta inmune y supera los mecanismos del organismo para combatir el cáncer (Nieto, 2011) e incluso a otro tipo de patologías como las infecciones, ya que estas mismas rutas son activadas tras una infección por patógenos (Hofman y Vouret-Craviari, 2012).
Mecanismos de regulación de la EMT: miRNAs
Para la EMT se han descrito otros niveles de modulación que incluyen las regulaciones epigenética y post-transcripcional (Nieto, 2011). Entre los mecanismos de regulación post-transcripcional, se encuentra la acción de los microRNAs (miRNAs), que ya han sido muy bien descritos en la EMT (Nieto et al., 2016; Cursons et al., 2018).
Los miRNAs son estructuras de aproximadamente 22 nucleótidos conocidos como reguladores postranscripcionales, ya que se asocian a proteínas argonautas (AGO) y forman un complejo de silenciamiento de RNA interferencia (RISC), el cual degrada o evita la traducción del mRNA blanco. Están involucrados en muchos procesos fisiológicos de la célula, como la división celular, la diferenciación, la apoptosis e incluso el desarrollo (Karim et al., 2016). Los miRNAs proveen una retroalimentación en la regulación de la EMT inducida por factores de crecimiento (Qin et al., 2016) y tanto miRNAs virales como celulares pueden mediar el potenciamiento o represión, respectivamente, del ciclo infeccioso del virus, lo cual los hace actores importantes a evaluar (Cullen, 2009; Tang et al., 2018).
Las familias de miRNAs reguladores de la EMT incluyen miR-200, miR-203 y miR-183, las cuales se han involucrado en la represión de la EMT al actuar sobre SNAIL1/2, Notch, ZEB1/2 y E-cadherina, entre otros (Hao et al., 2014). Las familias de los miR-200 (miR-200a/b/c) y de miR-34 (Dong et al., 2016) se han involucrado fuertemente en la represión de los procesos cancerígenos. Se han encontrado en distintos tipos de cáncer reprimiendo el factor de transcripción ZEB2, a SMAD y al TGFβ (Hao et al., 2014; Zaravinos, 2015). Así mismo, la activación de ZEB2 reprime la actividad de los miR-200 (Bullock et al., 2012). En el caso de miR-203, este reprime principalmente la proteína SNAI1; de manera opuesta, se ha observado que la expresión de miR-203 es reprimida por SNAI1 en células de cáncer de pulmón (Hao et al., 2014). Estos reguladores son positivos en la transcripción de E-cadherina y de inhibidores de la expresión de vimentina, lo cual reprime la EMT (Figs. 1 y 2b).
Por otro lado, en el panorama de las infecciones virales se han descrito miRNAs celulares sobre-expresados e implicados en vías moleculares relacionadas con la EMT. Este es el caso del miR-21-5p, el cual es también un oncomiR bien estudiado (Wang et al., 2011; Leidinger et al., 2014), regulador clave de procesos oncogénicos (Selcuklu et al., 2009) que, en respuesta a la infección por Coxsackievirus B3, rompe las uniones cadherina-catenina y desmosomas y altera la unión célula-célula (Ye et al., 2014). También se ha descrito que contribuye a la EMT inducida por CDK5 (Cycline dependent kinase) y TGF-β1 (Wang et al., 2014; Ren et al., 2015) y promueve la diferenciación, la migración y la invasión (Qin et al., 2016).
Los miR-146a y miR-146a-5p comparten la misma región semilla y se inducen en respuesta a una variedad de citoquinas y componentes microbianos. Se han encontrado en mujeres como partícipes de la sub-regulación de la expresión de BRCA1 (gen regulador de la proliferación celular) promoviendo la progresión del cáncer de seno (Garcia et al., 2011).
Se sabe que los miR-146a/b y miR-21 son inducidos por pre-miRNAs expresados por el EBV (Epstein Barr Virus) durante su infección (Skalsky et al., 2012). Además, el miR-484 se ha encontrado significativa y diferencialmente expresado en suero de pacientes con cáncer de seno en estado temprano, en comparación con pacientes sanos (Hu et al., 2012; Zearo et al., 2014); también se ha asociado a quimiorresistencia y modulación de angiogénesis (Vecchione et al., 2013).
Coincidencialmente, en un trabajo previo de nuestro grupo, se encontró que entre las estructuras similares a microRNAs predichas por biología computacional, el blanco celular con mayor puntaje y que además era único entre los ocho miRNAs del genoma del Dengue Virus (DENV) (Ospina-Bedoya et al., 2014), fue el receptor 2 de aquel ligando (FGFR2). Mediante el enriquecimiento de ontología de genes, se mostró además que podría explicar el fenotipo celular alterado en las infecciones virales.
PATÓGENOS VIRALES INDUCTORES DE LA EMT Y PATOGÉNESIS VIRAL
La infección por patógenos extra e intracelulares puede desencadenar las vías de señalización anteriormente mencionadas que convergen en la inducción de la EMT. Tanto factores de crecimiento extracelulares, como patógenos que son invasores intracelulares, comparten la activación de las mismas vías, siendo lógico que ciertos patógenos pueden ser inductores de la EMT (Thomas y Banks, 2018). Como refuerzo a esta idea, es bastante relevante el hecho de que exista una predisposición a los procesos fisiológicos relacionados con la EMT tras una infección bacteriana (Clarke et al., 2011) o viral (Hofman y Vouret-Craviari, 2012; Chen et al., 2016).
Se considera que los patógenos invasores usan el proceso de la EMT para facilitar su llegada o entrada (Clarke et al., 2011) a través de los epitelios e iniciar la infección. En los virus, como se mencionó al inicio, se han descrito algunos como inductores de la EMT. El virus Epstein-Barr ó Herpesvirus Humano 4 (de Oliveira et al., 2016), que además de células del sistema inmune también infecta epitelios, se ha reportado como promotor de carcinomas al activar a NF--SB y Erk1/2 (Lin et al., 2018) e inducir la EMT vía Akt/Erk y Syk/Src (Park et al., 2014). El virus del sarcoma de Kaposi o Herpesvirus Humano 8 (Gaur et al., 2019) puede promover la invasión al activar las MMPs, la angiogénesis (He et al., 2012) y la EMT, activando la señalización de Notch (Gasperini et al., 2012). Los virus de la Hepatitis B (HBV) y C (HCV) (Chargui et al., 2011; Hu et al., 2017) pueden inducir enfermedades crónicas que promueven el desarrollo de cirrosis y hepatocarcinoma (Bose et al., 2012). Se ha descrito que la proteína core del HCV interactúa con varias proteínas celulares en la inducción de la EMT (Tiwari et al., 2015), al igual que la proteína HBx (Hepatitis B-X Protein) del HBV (Ahodantin et al., 2019; Rawal et al., 2019). También se conoce que el Virus Ébola modula la señalización del TGF y de los marcadores mesenquimatosos en hepatocitos (Kindrachuk et al., 2014), entre otros (Xia et al. 2017). En el caso del virus del papilloma humano (VPH), aunque está fuertemente involucrado en la activación de cáncer uterino, aún no se ha reportado como un inductor de la EMT.
Estos hallazgos evidencian que las infecciones virales no sólo activan las mismas vías involucradas en la EMT (Fig. 1), sino que son capaces de predisponer al organismo a desarrollar metástasis. En el caso del DENV, aunque aún no se ha demostrado que sea inductor de la EMT, existe evidencia que parece insinuarlo (Jhan et al., 2017); la patogénesis de la enfermedad que causa está relacionada con la pérdida de permeabilidad vascular y la extravasación de plasma, lo que da indicios de que el DENV podría proponerse próximamente como inductor de la EMT.
En nuestro grupo de investigación hemos descubierto que la infección con DENV en células endoteliales produce un remodelamiento microvascular, lo cual se hizo mediante un abordaje de genómica no codificante (para extraer y cuantificar microRNAs), además de cuantificar citoquinas, quimioquinas y factores de crecimiento en el sobrenadante de cultivos celulares (Álvarez-Díaz et al., 2019). En este caso, podríamos postular que el DENV induce una EnMT (Endothelial-to-Mesenchymal Transition) porque el tipo celular que está bajo estudio proviene de líneas celulares de microvasculatura humana (HMEC-1) (ATCC, 2019).
De otra parte, está ampliamente demostrado que las cascadas de señalización con supervivencia celular están implicadas en la EMT (Tiwari et al., 2012). Así que no resultaría extraño que el DENV induzca tal tipo de señalización como un preámbulo a su efecto sobre la plasticidad celular. Hemos demostrado que DENV usa las señalizaciones PI3K y Akt para establecer y consolidar su ciclo replicativo (Cuartas-López et al., 2018). Además, tal señalización conduce a una activación de RhoGTPasas que, corriente abajo mediante sus efectores, son las responsables de la remodelación del citoesqueleto de actina. Tal fenómeno podría considerase como uno de los estados de la plasticidad que aún queda por demostrar. Adicionalmente, nuestro grupo encontró recientemente que CDK5 potencialmente participa en la consolidación de la infección por DENV (Roa-Linares y Gallego-Gómez, 2019), el cual es uno de los actores moleculares activadores de EMT mencionados anteriormente. Evidencias claras de la estrecha relación entre la infección por DENV y la activación de EMT.
CONCLUSIÓN
Desmantelar poco a poco las múltiples interacciones entre virus y hospederos contribuirá a mejorar nuestro entendimiento de la biología celular de la infección viral, así como en la propia biología del virus dengue en este caso. Este nuevo conocimiento permite además, afrontar los retos que aún existen en el control de enfermedades causadas por virus, así como epidemias y emergencias virales. En el caso particular del DENV, estos hallazgos poseen una pertinencia social muy alta en nuestro país debido a la patogénesis de la enfermedad que causa, la cual puede llegar a ser letal, lo que ya ha sido descrito por la Organización Mundial de la Salud (WHO, 2009). Las perspectivas del panorama actual van encaminadas a fortalecer la estrategia de Antivirales dirigidos a blancos celulares o HTA (Host-targeting antivirals) (Sayce et al., 2010) que, de la mano con las tecnologías para imagenología de células vivas actualmente desarrolladas, representan una herramienta clave con gran potencial en la evaluación de posibles agentes terapéuticos.

