










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Salud Uninorte
Print version ISSN 0120-5552On-line version ISSN 2011-7531
Salud, Barranquilla vol.26 no.2 Barranquilla July/Dec. 2010
ARTÍCULO DE REVISIÓN/ REVIEW ARTICLE
REVISIÓN BÁSICA/BASIC REVIEW
Estudios de asociación mediante rastreo genómico y su contribución en la genética del asma
Genome-wide association studies (gwas) and its contribution to the genetic of asthma
Yosed Anaya Chavez 1 Beatriz Martinez 2
1Estudiante de Maestría en Inmunología, Universidad de Cartagena. yosedpatricia@yahoo.es
2 Msc. Profesor asociado, Universidad de Cartagena. Jefe del Laboratorio de Genética Molecular del Instituto de Investigaciones Inmunológicas de la misma universidad.
Correspondencia: San Diego, Calle de la Tablada 7-57, Cartagena (Colombia). Fax: 6648053. beatri23@yahoo.com
Fecha de recepción: 29 de abril de 2010Fecha de aceptación: 30 de junio de 2010
Resumen
A pesar de todos los esfuerzos realizados por más de una década, las bases genéticas de muchas enfermedades comunes y complejas aún siguen siendo desconocidas, sin desmeritar los notables avances que se han logrado con los estudios de ligamiento en familias y de asociación con genes candidatos. Recientemente, el desarrollo de metodologías más robustas, como los estudios de asociación con rastreos genómicos (GWAs), ha permitido replicar asociaciones ya reportadas y a la vez descubrir nuevos genes posiblemente asociados. Los GWAs se basan en la utilización de un número considerable de marcadores genéticos tipo SNPs o STRs, los cuales son detectados con el propósito de encontrarlos asociados con la aparición y/o desarrollo de ciertas enfermedades. Teniendo en cuenta el gran impacto que actualmente tienen los GWAs como herramienta genética en la búsqueda de asociaciones, se hace una revisión teórica acerca del diseño e interpretación de los resultados de los mismos y su contribución en el asma y fenotipos relacionados.
Palabras clave: GWAs, asma, enfermedades complejas, IgE, marcadores genéticos, SNPs.
Abstract
Despite all the efforts of more than a decade, the genetic basis of many common and complex diseases are still unknown, without demerit the remarkable progress that has been made with the linkage studies in families and association studies of candidate genes. Recently, development of more robust methodologies, like genome-wide associations studies (GWAs), has allowed to replicate previously reported associations and at the same time discovers new possibly associated genes. The GWAs are based on the use of a considerable number of genetic markers like SNPs or STRs which are searched in order to of associate them with the appearance or development of certain diseases. Given the large current impact of the GWAs as genetic tool in the search of associations this is a theoretical review on the design and interpretation of results and contribution of GWAs in asthma and related phenotypes.
Key words: GWAs, Asthma, IgE, Genetics markers, Complex diseases, SNPs.
INTRODUCCIÓN
El asma es una enfermedad compleja que resulta de la interacción de diversos factores genéticos y ambientales. En los últimos años se ha observado un incremento en el descubrimiento de genes relacionados con dicha enfermedad; cerca de unos 100 loci para más de 40 enfermedades; los cuales han podido replicarse actualmente a través de herramientas más robustas como son los estudios de asociación con rastreos genómicos (GWAs) [1, 2].
GWAs es una técnica muy revolucionaria que permite investigar numerosas regiones del genoma humano y detectar un número considerable de marcadores genéticos tipo SNPs o STRs a niveles de resolución que anteriormente eran inalcanzables. A través de ellos se han identificado asociaciones estadísticamente significativas entre cientos de genes y enfermedades complejas comunes, lo cual ha contribuido con el incremento del conocimiento sobre las bases moleculares implicadas principalmente en la predisposición a padecerlas, como es el caso de la diabetes tipo I [3] y tipo II [4-7], enfermedad inflamatoria del intestino [8], cáncer de próstata [9, 10] [11, 12] y cáncer de seno [13] [14], asma [2, 15-21], enfermedad coronaria [22], fibrilación auricular [23], tuberculosis [24], artritis reumatoide [25-27], esquizofrenia [28], entre otras.
Teniendo en cuenta el gran impacto que actualmente tienen los GWAs como herramienta genética en la búsqueda de asociaciones con asma y fenotipos relacionados, ésta es una revisión rigurosa y teórica acerca del diseño e interpretación de los resultados de estos estudios y, de igual manera, su contribución en el asma y/o fenotipos relacionados.
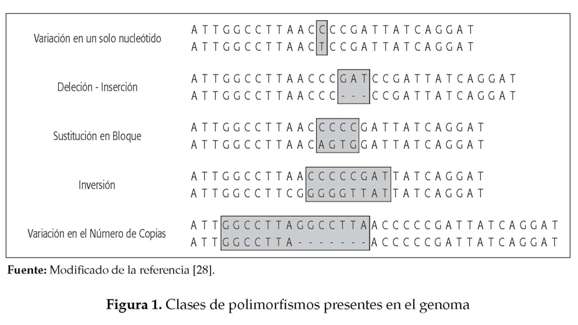
BREVE DESCRIPCIÓN DE LAS CLASES DE POLIMORFISMOS EN EL GENOMA HUMANO
Si bien dos personas del mismo sexo comparten un porcentaje elevado (alrededor del 99,9%) de su secuencia de ADN, el 0.1% restante contiene las variaciones genéticas que influyen en el fenotipo de los individuos. Descubrir las variantes que contribuyen al riesgo de una enfermedad común ofrece una de las mejores oportunidades para la comprensión de las causas de dicha enfermedad en los seres humanos. Seguidamente se describirán de manera breve los diferentes polimorfismos o variantes que se encuentran en el genoma humano.
Las variaciones en el genoma se clasifican principalmente en variantes comunes y variantes raras para indicar la frecuencia del alelo menor presente en la población. Las comunes son polimorfismos definidos como aquella variante genética cuya frecuencia del alelo menor (MAF) es de por lo menos un 1% en la población, mientras que las variantes raras son aquellas que tienen una MAF menor del 1%. De igual forma, las variantes en el genoma se pueden clasificar de acuerdo con su composición de nucleótidos en variantes de un solo nucleótido y variantes estructurales. La gran mayoría de las variantes son neutras, decir que no contribuyen a la variación fenotípica [29].
Los polimorfismos en un solo nucleótido (SNPs) son las más frecuentes en los individuos; se ha estimado que el genoma contiene alrededor de 11 millones, de los cuales 7 millones aproximadamente con un MAF del 5% y los restantes con un MAF entre 1 y 5%. Por su parte, las variantes estructurales se refieren a secuencias de pares de bases que difieren entre los individuos, como son las inserciones-dilecciones (Andes). Existen además dos tipos de polimorfismos que corresponden a sustituciones en bloque, en las cuales un segmento de nucleótidos adyacentes varía entre dos genomas. También se encuentran las inversiones de secuencias de ADN, donde pares de bases son invertidas en una sección de un cromosoma, y por último, las variaciones en el número de copias (CNV), que son secuencias idénticas o casi idénticas que se repiten en algunos cromosomas pero no en otros [29]. En la figura 1 se resumen las clases de variantes descritas previamente. En adelante nos enfocaremos en los SNPs, ya que son los polimorfismos más frecuentemente utilizados en los estudios de GWAs.
En la actualidad, los GWAs son las herramientas más utilizadas en las investigaciones genéticas. A través de ellos se genotipifican grupos de SNPs para encontrar variantes asociadas con enfermedades; también se identifican rasgos cuantitativos heredables que se constituyen como factores de riesgo para el desarrollo de las patologías. Todos los SNPs utilizados como marcadores en estos estudios han sido registrados en las bases de datos del Centro Nacional de Información Biotecnológica (NCBI), incluyendo sus frecuencias alélicas y otro tipo de información genómica [30].
A través de los GWAs se pueden detectar también las interacciones existentes entre genes, las modificaciones de las asociaciones de una variante genética por otra, como sucede en la enfermedad de Alzheimer con los genes APOE y GAB2 [31]; además, se pueden detectar haplotipos de alto riesgo o combinaciones de múltiples SNPs dentro de un único gen, como ocurre en la Fibrilación Auricular [23]. Uno de los principales aportes al desarrollo de esta herramienta son: primero, los resultados obtenidos a partir del proyecto HapMap, los cuales muestran los patrones de variación y de desequilibrio de ligamiento de tres poblaciones ancestrales como son la caucásica, asiática y africana [32], y segundo, la disponibilidad de micro chips de genotipificación que agrupan cientos de miles SNPs, los cuales abarcan gran parte del genoma total y favorecen la realización de un rastreo global.
Desde sus inicios, los GWAs han utilizado marcadores moleculares del tipo STRs o SNPs. Con respecto a los STRs, el genoma humano está constituido por aproximadamente 30,000 microsatélites, los cuales fueron descritos inicialmente por Weber y cols. [33] como secuencias pequeñas de ADN repetidas en tándem (STRs), constituidas por unidades de 2 a 7 pb que se repiten en un número variable de veces y dan lugar a alelos de tamaño aproximado entre 80 y 400 pb. Su gran utilidad se puso rápidamente de manifiesto como marcadores moleculares para rastrear genes comprometidos en enfermedades, debido a su abundancia, distribución muy regular en el genoma, naturaleza polimórfica y su pequeño tamaño.
Los microsatélites son altamente polimórfi-cos, tienen un alto grado de heterozigosidad (70% aproximadamente) y la longitud de su desequilibrio de ligamiento (LD) está en el rango de 100kb comparado con el que presentan los SNPs (aproximadamente 30kb) [25, 34]. Debido a esto, los STRs tienen la ventaja de abarcar una extensa región genómica y pueden ser utilizados en pequeño número para analizar asociaciones en el genoma. Asimismo, los microsatélites muestran un alto contenido de información genética, es decir que alrededor de seis a diez alelos pueden ser evaluados [34].
Por su parte, los SNPs, que como se describió corresponden a una variación en una sola base (adenina (A), timina (T), citosina (C) o guanina (G)) en una secuencia del genoma y pueden originarse por transiciones o transversiones. En las transiciones es sustituida una base por otra que pertenece a la misma categoría química (una purina por otra purina o una pirimidina por otra pirimidina), mientras que en las transversiones ocurre lo contrario (una purina por una pirimidina y viceversa). Actualmente, más de 11 millones de esta clase de polimorfismos se han depositado en las bases de datos publicadas, la mayoría de los cuales no presentan efecto funcional [34].
Los SNPs pueden estar presentes en regiones codificadoras o no codificadoras, así como en regiones intergénicas del genoma. Inicial-mente, estos polimorfismos se observaron en regiones que codifican genes indispensables para la expresión de proteínas. Los SNPs que se encuentran al inicio de regiones que codifican proteínas pueden ejercer efectos sobre varios eventos epigenéticos, splicing genético, sitios de unión de los factores de transcripción, regulación de promotores y RNAs no codificantes [34].
El aspecto más importante con respecto al uso de estos marcadores en los estudios de rastreo genómico es que ellos se pueden heredar juntos en un mismo haplotipo o bloque de LD. Por consiguiente, para la genotipificación se puede emplear uno o un grupo de estos marcadores presentes en una región del genoma, ya que todos los SNPs que se encuentran en ese bloque están relacionados. Sin embargo, en una región del genoma que tenga poco LD o donde no existan segmentos de haplotipos se necesitaría un número mayor de SNPs [34].
En cuanto al estudio de variantes raras, éstas no pueden ser detectadas a través de GWAs que se basan en el uso de marcadores poli-mórficos que están ligados, debido a que son variantes de baja frecuencia y pequeña contribución en la susceptibilidad a enfermedades. Por lo tanto, para establecer asociaciones con variantes raras es necesario realizar un mapeo directo e identificar dichas variables dentro de la población de estudio. La identificación se realiza mediante la secuenciación de genes candidatos en el grupo de individuos con la enfermedad en cuestión. Posteriormente se evalúa su frecuencia en una población control apropiada y las posibles consecuencias en la función del producto de dicho gen [35, 36].
TIPOS DE DISEÑOS PARA LA REALIZACIÓN DE GWAS
El diseño del estudio que se utiliza frecuentemente en los GWAs es el de casos y controles, en el cual las frecuencias alélicas de los individuos con la enfermedad de interés son comparadas con la del grupo control (personas sin la enfermedad). Este tipo de diseño se caracteriza porque los participantes de ambos grupos del estudio (casos y controles) son seleccionados de la misma población; además, la muestra de los pacientes debe ser representativa con relación a todos los casos de la enfermedad y los datos epidemiológicos y genéticos son recolectados de forma similar en los casos, así como en los controles. Este diseño tiene la ventaja de ser rápido y económico, sin embargo, es muy sensible a los sesgos sistemáticos, como es el sesgo recall y el sesgo de selección [34, 37, 38].
Otro tipo de diseño empleado en los rastreos genómicos es el estudio de cohorte, en el cual los datos de exposición y las muestras de ADN son obtenidos de un grupo de individuos que no padece la enfermedad. Estas personas son monitoreadas a través de un seguimiento hasta que la enfermedad de interés aparezca o se desarrolle en ellos. Los individuos que participan en este tipo de estudio son más representativos de la población de donde fueron seleccionados. Además, la enfermedad o el rasgo de interés son verificados igualmente en individuos con o sin la variabilidad en el genoma. Los casos son incidentes, es decir, se desarrollan durante el período de observación. La desventaja principal de estudios de cohorte es la gran inversión de tiempo y su costo. Se requiere una amplia cohorte y el seguimiento a través del tiempo para detectar los efectos genéticos moderados de los genes en estudio [34, 37, 38].
Los Diseños basados en Familia (FABT) son un tipo especial de estudios en los que se analizan solamente los casos de individuos afectados y sus parientes. Sólo la descendencia afectada es incluida en el estudio, sin embargo, la genotipificación es llevada a cabo en los tres miembros de la familia. En este tipo de estudio se estima la frecuencia con la cual un alelo es transmitido a la descendencia afectada por los padres heterozigóticos [37].
De igual forma, muchos estudios de GWAs utilizan diseños de varias etapas (multistage designs) con el fin de reducir el número de resultados falsos positivos, disminuir los costos y mantener un poder estadístico adecuado. Inicialmente el estudio se desarrolla en un grupo de casos y de controles, donde se evalúa un grupo de SNPs asociados, luego, los SNPs que detecten variabilidad en el genoma son replicados en un segundo y posteriormente en un tercer grupo de casos y controles [37]. Actualmente es el diseño de excelencia.
PLATAFORMAS COMERCIALES DE USO EN GWAS
Dentro de las metodologías iníciales empleadas en la genotipificación de las regiones variables en el genoma se encuentran los análisis con enzimas de restricción (RFLP), amplificación específica de alelos (PCR), extensión de una base simple, ensayos basados en ligamiento, hibridación oligonucleotídica específica de alelo y un sinnúmero de métodos basados en electroforesis [37, 39].
Estas técnicas han sido incorporadas dentro de una amplia variedad de plataformas comerciales con algunas modificaciones a los métodos generales que han sido descritos anteriormente. Sumado a lo anterior, el desarrollo tecnológico de microchips que contienen un número considerable de SNPs permite hacer mediciones rápidas de una gran cantidad de fenotipos en los individuos.
Actualmente, dentro de las plataformas ampliamente utilizadas en estos estudios se encuentran la plataforma de Affymetryx GenChip y la plataforma de Illumina BeadChip [34, 40]. Ambas plataformas han diseñado microchips que brindan información sobre la variación en el número de copias encontrada en el genoma. Affymetrix ofreció por primera vez una plataforma de alto rendimiento con su producto de 100K, que consiste de dos matrices capaces de genotipificar alrededor de 50,000 SNPs cada una, logrando así un total de 100.000 SNPs; esta plataforma ofrece el doble de la capacidad para localizar el gen implicado en la susceptibilidad a la enfermedad en cuestión. Esta casa comercial también ofrece una plataforma de 500K que permite la genotipificación de 500,000 SNPs [34] (ver tabla 1).
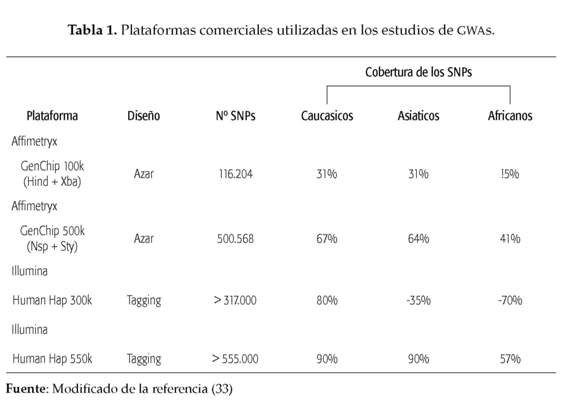
Los SNPs comprendidos en la matriz de 500K fueron seleccionados de las bases de datos de SNPs de dominio público (NCBI) y de Perlegen basadas en la frecuencia de ocurrencia, el tipo y análisis de desequilibrio de ligamiento (LD) en las poblaciones caucásicas, afroamericanas y asiáticas. El promedio de heterogozidad de los SNPs es de 0.30 y la distancia promedio entre ellos es de 5.8kb. Además de las pruebas con cada SNP, Affymetrix ha incluido un número de pruebas control dentro de los "arrays" con el propósito de asegurar la calidad de los resultados [34].
Por otra parte, la casa comercial Ilumina ha incorporado la tecnología de los BeadArray (análisis con microesferas) a la línea de productos ofrecidos BeadChip. Mientras los microarrays convencionales son creados mediante la unión de sondas o síntesis de sondas in situ, los BeadArray utilizan el au-toensamblaje al azar de un grupo de microes-feras dentro de un sustrato estandarizado. Cada microesfera (3um) es recubierta con sondas de secuencia específicas y los lotes de estas microesferas son ensamblados en los micropozos del array. Para determinar cuál microesfera se encuentra en cada micro pozo, oligonucleótidos marcados con fluorescencia son hibridizados a un segmento de las sondas conocido como secuencia de direccionamiento [34].
Los BeadChips de Ilumina utilizados en los estudios de GWAs son de 300 y 550K; ellos ofrecen cerca de 317.000 y 555.000 tagSNPs respectivamente. De igual forma, estos productos hacen uso de los ensayos Infinium de Ilumina; este tipo de ensayo le ha permitido a IIlumina ofrecer pruebas que arrojen la máxima información sobre los tagSNPs. Los tagSNPs presentes en el Chip de 300k fueron seleccionados de la primera fase del proyecto HapMap, mientras los SNPs del chip 550K se escogieron de ambas fases de este proyecto [34].
SELECCIÓN DEL NÚMERO DE MARCADORES SNPS Y TAMAÑO MUESTRAL
La mayoría de los marcadores utilizados en los estudios GWAs se localizan dentro de segmentos que presentan fuerte desequilibrio de ligamiento [41], es decir, están fuertemente correlacionados entre sí y los cromosomas contienen sólo algunos haplotipos en común. Actualmente, varias regiones genómicas (de aproximadamente 500kb) han sido minuciosamente examinadas como parte del proyecto ENCODE (Biblioteca de elementos de DNA). En este proyecto se resecuenciaron 96 cromosomas para determinar todas las variantes comunes, y además se genotipificaron todos los polimorfismos en un solo nucleótido que se encontraban en las bases de datos publicadas o que ya habían sido resecuenciados en otros estudios. Estos estudios muestran que la mayoría de los 11 millones de SNPs comunes en el genoma están agrupados y correlacionados unos con otros, al igual que el genotipo de uno de ellos puede predecir perfectamente el genotipo de su vecino. Teniendo en cuenta lo anterior, un SNPs puede representar muchas variantes de este tipo en un estudio de asociación, lo que se conoce como tagSNPs.
Una vez conocidos los patrones de Desequilibrio de Ligamiento (LD) de una región dada, se pueden elegir algunos tagSNPs a partir de los cuales se puede detectar la mayoría de las variantes comunes dentro de la región de interés.
ANÁLISIS DE LOS RESULTADOS DE GWAS
Las asociaciones con los alelos de cada SNP se hacen comparando la frecuencia de cada alelo en los casos y los controles. También se pueden comparar las frecuencias de los 3 genotipos resultantes en ambos grupos. Los análisis también pueden incluir el modelo genético (dominante, recesivo y aditivo), siendo el modelo aditivo el más usado en estos estudios. Los odds ratios son también calculados, y generalmente están en el rango de 1.2 a 1.3, lo que constituye un riesgo modesto [37].
La mayoría de estos estudios también calculan el factor de riesgo poblacional, pero tales estimados son casi siempre elevados, debido a que los odds ratios sobrepasan el riesgo relativo requerido para calcular dicho factor. Este estimado inicial exagerado lleva con mucha frecuencia a estudios de replicación pero que carecen de un tamaño de muestra suficiente y poder para replicar la asociación, debido a que se necesita un gran tamaño de la muestra para obtener odds ratios más pequeños [37].
La complejidad de los análisis se debe a que como es un estudio de múltiples pruebas, las asociaciones deben repetirse para 100000 o 1 millon de SNPs probados. La p convencional, es decir, menor de 0.05, en estudios de asociación de 1 millón de SNPs mostrarán alrededor de 50000 SNPs asociados a la enfermedad, de los cuales casi todos son falsos positivos. Por esto es necesario aplicar correcciones como la de Bonferroni, en la cual los valores de p convencional se dividen por el número de SNPs analizados. En un rastreo de 1 millón de SNPs se debe usar un umbral de p<0.05/106. Esta corrección ha sido objeto de controversia, debido a que asume asociaciones independientes de cada SNP con los fenotipos de la enfermedad, sabiendo que teóricamente dichos SNPs están correlacionados en algún modo debido al LD [37, 42].
Se han propuesto otros tipos de análisis que incluyen el estimado de la tasa de falsos positivos, la probabilidad del reporte de falsos positivos, la probabilidad de que la hipótesis nula sea verdadera, dando una significancia estadísticamente significativa y el estimado de los factores del teorema de Bayes [43, 44], es decir, incorporar la probabilidad a priori de la asociación basada en las características de la enfermedad o del SNP específico [3, 45]. Hasta la fecha la corrección de Bonferroni es la más usada para comparaciones múltiples en los estudios de GWAs [37].
Con el fin de lograr una mayor confiabilidad en las asociaciones encontradas a partir de los estudios de GWAs, es necesario excluir todos aquellos datos que representan falsos positivos, los cuales son producto de la estratificación poblacional que puede existir entre el grupo de casos y controles. Actualmente existen diversos métodos estadísticos que se aplican con el fin de identificar y corregir este fenómeno, los cuales se fundamentan en el análisis de un grupo de marcadores de control genético (Microsatélites y Marcadores Informativos de Ancestría) que están distribuidos a través del genoma y no son marcadores candidatos de asociación. El proyecto Hap-Map proporciona frecuencias alélicas para millones de SNPs presentes en las poblaciones continentales, que pueden ser utilizados para detectar la estratificación en poblaciones mezcladas como áfro-americanas, latinas o hispanas [46, 47].
LIMITACIONES EN LOS ESTUDIOS GWAS
Estos estudios muestran varias limitaciones. Los resultados falsos positivos, la falta de información sobre la función de un gen, la incapacidad para detectar variantes raras y estructurales, los posibles sesgos debido a la mala selección de los casos y controles, al igual que errores en la genotipificación, son los problemas más frecuentes en estos estudios. Además de lo anterior, es difícil identificar a través de dichos estudios las interacciones entre gen-ambiente, ya que existe una limitada información disponible sobre la exposición ambiental y otros factores de riesgo no genéticos [37].
La replicación de los resultados obtenidos a través de los GWAs es una de las estrategias más confiables con la cual se logra diferenciar las asociaciones que corresponden a falsos positivos de aquellas que son verdaderos positivos. No obstante, la reproducibilidad de estos estudios es complicada por factores como la estratificación de la población [48-50], diferencias fenotípicas, sesgos de selección, errores de genotipificación, entre otros. Actualmente, la mejor forma de evitar estas inconsistencias es hacer los estudios de replicación con un gran tamaño de muestra [34, 37].
A través de los GWAs se generan hipótesis, por lo tanto, la replicación es esencial en estos estudios para confirmar los resultados y evidenciar las asociaciones genéticas autenticas. Por otra parte, se hace necesario demostrar el efecto biológico del gen asociado a la enfermedad mediante pruebas funcionales in vivo o in vitro, replicables en modelos animales, para ratificar el papel significativo del gen detectado en la patogénesis de la enfermedad [51].
GWAS EN ASMA
El asma es una enfermedad de etiología desconocida, caracterizada por una inflamación intermitente de las vías respiratorias, la cual conduce con el tiempo a una obstrucción de la misma, inflamación crónica con eosinofília e hipertrofia de las células musculares lisas bronquiales.
Hace parte del repertorio de enfermedades complejas y su incidencia está determinada por la interacción de factores genéticos y ambientales. Además, es una enfermedad multifactorial y poligénica, lo que explica en parte su patrón de herencia complejo, diferente del de las enfermedades monogénicas, en las que se puede establecer con relativa facilidad, por ejemplo, si el fenotipo es dominante o recesivo, el grado de penetrancia,etc. [52, 53].
Durante mucho tiempo esta enfermedad ha sido objeto de gran interés para la comunidad científica, ya que sus bases moleculares no se han podido esclarecer totalmente. Por consiguiente, se realizan hasta la fecha muchos esfuerzos con el propósito de identificar genes cuyas variantes influyan notablemente sobre la patogénesis de la enfermedad.
La búsqueda de los genes relacionados con la etiología del asma se ha basado en el uso de diversas estrategias de epidemiología genética, apoyadas últimamente por los resultados del Proyecto Genoma Humano y el proyecto HapMap. Dentro de los estudios aplicados para alcanzar tal fin se destacan los estudios de ligamiento genético realizados en familias, las asociaciones de genes candidatos, donde se tiene información previa del gen que va a ser objeto de investigación, y los estudios más recientes conocidos como estudios de asociación con rastreo genómico [2, 17, 54-57].
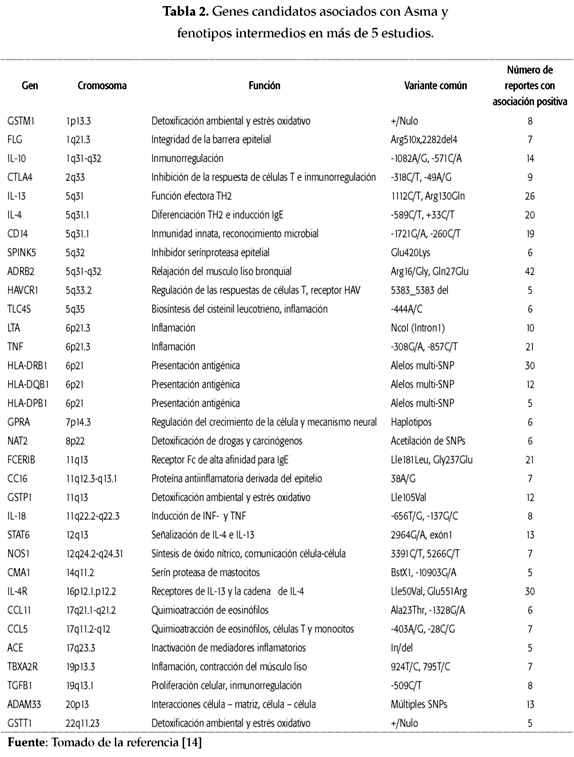
Gracias al incremento de las herramientas moleculares, actualmente existe un amplio registro de genes que están implicados en el desarrollo del asma. Recientemente, Vercelli y cols. [2] a través de estudios con genes candidatos clasificaron los genes de susceptibilidad a asma en cuatro categorías principales: los asociados con la respuesta inmune e inmunoregulación, genes asociados con la diferenciación de las células Th2 y funciones efectoras, genes asociados con la biología epitelial y la inmunidad a mucosas y finalmente genes asociados con la función pulmonar, remodelamiento de las vías aéreas y severidad de la enfermedad. Además, identificaron 33 genes en cinco o más estudios independientes (ver tabla 2). En 2006 Ober y cols. [17] reportaron 118 genes asociados con fenotipos de atopia y asma, los cuales habían sido replicados por lo menos una vez en diferentes poblaciones y revisados después por Vercelli D. [2].
Actualmente existen reportes de estudios de asociación con genes candidatos en la población colombiana, por medio de los cuales se observa una relación significativa entre polimorfismos en el gen GPR154 con asma y niveles de IgE total [58]; estos estudios han sido replicados en otras poblaciones como la caucásica y china. Por otra parte, Vergara y cols.[59] en estudios basados en familias encontraron que un haplotipo en el gen ADAM33 estuvo asociado con asma. Finalmente, Acevedo y cols.[60] en 2008 reportaron una asociación del SNPs C-509T en el gen TGF|3 con IgE total y específica para D. pteronyssinus en pacientes asmáticos.
Los estudios de GWAs en asma han descubierto nuevos genes candidatos con un efecto moderado sobre el fenotipo (ver tabla 3). Los resultados del primer estudio de GWAs en asma fueron publicados por Moffatt y cols. [15], quienes caracterizaron más de 317.000 SNPs en 499 pacientes con asma de la niñez y 1.243 individuos sanos, y encontraron que múltiples marcadores en el cromosoma 17q21.1 están fuertemente asociados con asma en la infancia. Estos resultados fueron replicados en 2.320 niños pertenecientes a una cohorte alemana y en 3.301 individuos de una cohorte británica de 1958. Es de anotar que se reporta un nuevo gen, OMRDL3, cuyos niveles de expresión son determinantes de susceptibilidad a asma en la niñez [15]. Estos estudios fueron replicados en una población franco-canadiense por Madore y cols. [18], quienes encontraron una fuerte asociación de 10 SNPs localizados en el cromosoma 17q21 con asma.
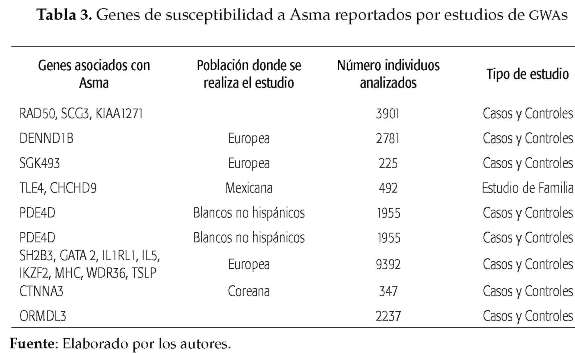
Estudios recientes realizados por J. Hui y cols. [16] utilizando 23.000 marcadores microsaté-lites identificaron en población australiana dos genes candidatos en el cromosoma 18 que posiblemente podrían controlar los niveles de expresión de la IgE. Además, demostraron la viabilidad de utilizar STRs en la metodología de los GWAs para identificar posibles genes candidatos en otras enfermedades.
Rogers y cols. [61] evaluaron la reproducibi-lidad de asociaciones con asma previamente reportadas y la contribución de la variación genética alrededor de estos loci en la susceptibilidad a asma. En este estudio se identificaron 10 SNPs en seis genes, los cuales están significativamente asociados con la enfermedad, incluyendo la replicación del gen integrina | 3 (ITGB3). Además, la evaluación de 619 variantes adicionales reveló la asociación de 15 genes con el fenotipo de asma, teniendo en cuenta que nueve de esos genes fueron corregidos por múltiples comparaciones.
Los GWAs han hecho posible asociar por primera vez variantes de genes implicados en la regulación de los niveles de IgE total en suero con atopia y asma. Weidinger y cols. [62] examinaron más de 11.000 individuos alemanes de cuatro cohortes basadas en estudios de poblaciones, en las cuales observaron la presencia de variantes funcionales en los genes que codifican la cadena a del receptor de la IgE (FCERIA) en el cromosoma 1q23 (rs2251746 y rs2427837), asociado fuertemente con los niveles de IgE total. Además, los resultados encontrados en esta investigación confirman la asociación de STAT6 con los niveles de esta Ig en suero y sugieren que variaciones en el gen RAD50 pueden representar factores adicionales dentro del grupo de genes de ci-toquinas en el cromosoma 5q3. Sin embargo, los autores resaltan la necesidad de continuar con las investigaciones en esta región.
Por su parte, Rafaels y cols. [63] en un rastreo realizado en una población de descendencia africana demostraron una fuerte asociación de los genes ace, spinks, nos1 y adam33 con asma, validando la asociación ya reportada anteriormente en otras poblaciones. Los resultados de esta investigación indican que varios de los genes replicados están fundamentalmente comprometidos en el desarrollo de la enfermedad a pesar de diversidad étnica existente.Adicionalmente, Y. J. Tsai y cols. [20] identificaron una fuerte asociación entre asma y el gen cul5 localizado en el cromosoma 11q22, que no ha sido estudiado anteriormente. Este hallazgo confirma una vez más que a través de los estudios de asociación con rastreos genómicos se pueden detectar nuevos genes que determinan la aparición de enfermedades complejas como el asma.
CONCLUSIONES
Los estudios de rastreo genómico son las herramientas moleculares más usadas actualmente para detectar variaciones genéticas que confieren susceptibilidad a enfermedades complejas.
• A través de ellos se han identificado nuevos genes comprometidos en el desarrollo de diversas enfermedades complejas. De igual forma, se han realizado estudios de replicación con genes anteriormente reportados en diferentes poblaciones.
• El diseño de multistage es el más utilizado, ya que reduce el número de falsos positivos, el costo del estudio y mantiene su poder estadístico.
Existe una gran variedad de plataformas comerciales disponibles para realizar los gwas ofrecidas por las casas comerciales Affimetrix e Illumina; la elección de una de ellas depende en gran medida del interés del investigador y de la población objeto de estudio, teniendo en cuenta que estas plataformas se basan en los resultados obtenidos por el proyecto HapMap realizado en tres poblaciones, como son caucásica, afroamericana y asiática, las cuales presentan diferencias en su estructura genética.
• Apesar de los grandes avances alcanzados hasta el momento por los gwas en relación con el descubrimiento de genes asociados a enfermedades, las interacciones entre estos genes y el ambiente no se han establecido, por lo que se hace necesario enfatizar las investigaciones acerca de este tema, el cual es fundamental para entender la etiología de las enfermedades complejas.
• Hasta el momento no se han realizado estudios funcionales que confirmen del papel significativo de los genes que han sido asociados a enfermedades complejas como el asma a través de los gwas, y además, los estudios de replicación en diferentes poblaciones ha sido complicado.
Conflicto de interés: Ninguno. Financiación: Universidad de Cartagena.
REFERENCIAS
1. Kabesch M. Novel asthma-associated genes from genome-wide association studies: what is their significance? Chest 137(4): 909-15. [ Links ]
2. Vercelli D. Discovering susceptibility genes for asthma and allergy. Nat Rev Immunol 2008; 8(3): 169-82. [ Links ]
3. Genome-wide association study of 14,000 cases of seven common diseases and3,000 shared controls. Nature 2007; 447(7145): 661-78. [ Links ]
4. Sladek, R. et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 2007; 445(7130): 881-5. [ Links ]
5. McCarthy M.I., Zeggini E. Genome-wide association scans for Type 2 diabetes: new insights into biology and therapy. Trends Pharmacol Sci, 2007; 28(12): 598-601. [ Links ]
6. McCarthy MI, Zeggini E. Genome-wide association studies in type 2 diabetes. Curr Diab Rep 2009; 9(2): 164-71. [ Links ]
7. Lyssenko V, Groop L. Genome-wide association study for type 2 diabetes: clinical applications. Curr Opin Lipidol 2009; 20(2): 87-91. [ Links ]
8. Duerr RH et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science, 2006; 314(5804):1461-3. [ Links ]
9. Thomas G. et al. Multiple loci identified in a genome-wide association study of prostate cancer. Nat Genet 2008; 40(3): 310-5. [ Links ]
10. Yeager M. et al., Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet 2007; 39(5): 645-9. [ Links ]
11. Xu J et al. Association of prostate cancer risk variants with clinicopathologic characteristics of the disease. Clin Cancer Res 2008; 14(18): 5819-24. [ Links ]
12. Kiemeney LA. Words of wisdom. Re: genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Eur Urol 2007; 52(3): 920-1. [ Links ]
13. Easton DF et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 2007; 447(7148): 1087-93. [ Links ]
14. Zheng W et al. Genome-wide association study identifies a new breast cancer susceptibility locus at 6q25.1. Nat Genet 2009; 41(3): 324-8. [ Links ]
15. Moffatt M F. et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature 2007; 448(7152): 470-3. [ Links ]
16. Hui J et al. A genome-wide association scan for asthma in a general Australian population. Hum Genet 2008; 123(3): 297-306. [ Links ]
17. Ober C., Hoffjan S. Asthma genetics 2006: the long and winding road to gene discovery. Genes Immun 2006; 7(2): 95-100. [ Links ]
18. Madore AM et al. Replication of an association between 17q21 SNPs and asthma in a French-Canadian familial collection. Hum Genet 2008; 123(1): 93-5. [ Links ]
19. Choudhry S et al. Genome-wide screen for asthma in Puerto Ricans: evidence for association with 5q23 region. Hum Genet 2008; 123(5): 455-68. [ Links ]
20. Y.J. Tsai et al. A Genome Wide Approach to Identify Genetic Determinants of Asthma Traits Related to Airway Function in Two Populations of African Descent Journal of Allergy and Clinical Immunology 2009;123(2): S148. [ Links ]
21. Willis-Owen SA, Cookson WO, Moffatt MF. Genome-wide association studies in the genetics of asthma. Curr Allergy Asthma Rep 2009; 9(1): 3-9. [ Links ]
22. McPherson R. et al. A common allele on chromosome 9 associated with coronary heart disease. Science 2007; 316(5830): 1488-91. [ Links ]
23. Gudbjartsson DF et al. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature 2007; 448(7151): 353-7. [ Links ]
24. Mahasirimongkol S et al. Genome-wide SNP-based linkage analysis of tuberculosis in Thais. Genes Immun 2009; 10(1): 77-83. [ Links ]
25. Tamiya G. et al. Whole genome association study of rheumatoid arthritis using 27 039 microsatellites. Hum Mol Genet 2005; 14(16): 2305-21. [ Links ]
26. Yamamoto K, Yamada R. Genome-wide single nucleotide polymorphism analyses of rheumatoid arthritis. J Autoimmun 2005; 25 Suppl: 12-5. [ Links ]
27. Wei Z, Li M. Genome-wide linkage and association analysis of rheumatoid arthritis in a Canadian population. BMC Proc 2007; 1Suppl 1: S19. [ Links ]
28. Kirov G et al. A genome-wide association study in 574 schizophrenia trios using DNA pooling. Mol Psychiatry 2008. [ Links ]
29. Frazer KA et al. Human genetic variation and its contribution to complex traits. Nat Rev Genet 2009; 10(4): 241-51. [ Links ]
30. Sayers EW et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 2009; 37(Database issue): D5-15. [ Links ]
31. Reiman EM et al. GAB2 alleles modify Alzheimer's risk in APOE epsilon4 carriers. Neuron 2007; 54(5): 713-20. [ Links ]
32. A haplotype map of the human genome. Nature 2005; 437(7063): 1299-320. [ Links ]
33. Weber JL, May PE. Abundant class of human DNA polymorphisms which can be typed using the polymerase chain reaction. Am J Hum Genet 1989; 44(3): 388-96. [ Links ]
34. D. C. Rao. C.C.G. Genetic Dissection of Complex Traits 2008; 60. [ Links ]
35. Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet;11(6): 415-25. [ Links ]
36. Bodmer W, Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet 2008; 40(6): 695-701. [ Links ]
37. Pearson TA, Manolio TA. How to interpret a genome-wide association study. JAMA 2008; 299(11): 1335-44. [ Links ]
38. Cordell HJ, Clayton DG. Genetic association studies. Lancet 2005; 366(9491): 1121-31. [ Links ]
39. Mamotte CD. Genotyping of single nucleo-tide substitutions. Clin Biochem Rev 2006; 27(1): 63-75. [ Links ]
40. Neale BM, Purcell S. The positives, protocols, and perils of genome-wide association. Am J Med Genet B Neuropsychiatr Genet 2008; 147B(7): 1288-94. [ Links ]
41. Kruglyak L. The road to genome-wide association studies. Nat Rev Genet 2008; 9(4): 314-8. [ Links ]
42. Yang Q et al. Power and type I error rate of false discovery rate approaches in genome-wide association studies. BMC Genet 2005; 6Suppl 1: S134. [ Links ]
43. Zhang Y, Liu JS. Bayesian inference of epistatic interactions in case-control studies. Nat Genet 2007; 39(9): 1167-73. [ Links ]
44. Wakefield J. Bayes factors for genome-wide association studies: comparison with P-values. Genet Epidemiol 2008; 33(1): 79-86. [ Links ]
45. McCarthy MI et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet 2008; 9(5): 356-69. [ Links ]
46. Tian C, Gregersen PK, Seldin MF. Accounting for ancestry: population substructure and genome-wide association studies. Hum Mol Genet 2008; 17(R2): R143-50. [ Links ]
47. Enoch MA et al. Using ancestry-informative markers to define populations and detect population stratification. J Psychopharmacol 2006; 20(4 Suppl): 19-26. [ Links ]
48. Teo YY. Common statistical issues in genome-wide association studies: a review on power, data quality control, genotype calling and population structure. Curr Opin Lipidol 2008; 19(2): 133-43. [ Links ]
49. Marchini J et al. The effects of human population structure on large genetic association studies. Nat Genet 2004; 36(5): 512-7. [ Links ]
50. Freedman ML et al. Assessing the impact of population stratification on genetic association studies. Nat Genet 2004; 36(4): 388-93. [ Links ]
51. Seng KC, Seng CK. The success of the genome-wide association approach: a brief story of a long struggle. Eur J Hum Genet 2008; 16(5): 554-64. [ Links ]
52. Caraballo LGE. Asma. Asthma 2005; 45 - 53. [ Links ]
53. Koppelman GH. Gene by environment interaction in asthma. Curr Allergy Asthma Rep 2006; 6(2): 103-11. [ Links ]
54. Mathias RA et al. A genome-wide association study on African-ancestry populations for asthma. J Allergy Clin Immunol; 125(2): 336-346 e4. [ Links ]
55. Li X et al. Genome-wide association study of asthma identifies RAD50-IL13 and HLA-DR/ DQ regions. J Allergy Clin Immunol; 125(2): 328-335 e11. [ Links ]
56. Sleiman PM et al. Variants of DENND1B associated with asthma in children. N Engl J Med; 362(1): 36-44. [ Links ]
57. Wu H et al. Evaluation of candidate genes in a genome-wide association study of childhood asthma in Mexicans. J Allergy Clin Immunol; 125(2): 321-327 e13. [ Links ]
58. Vergara C et al. Association of G-protein-coupled receptor 154 with asthma and total IgE in a population of the Caribbean coast of Colombia. Clin Exp Allergy 2009; 39(10): 1558-68. [ Links ]
59. Vergara CI et al. A Six-SNP haplotype of ADAM33 is associated with asthma in a population of Cartagena, Colombia. Int Arch Allergy Immunol; 152(1): 32-40. [ Links ]
60. Acevedo N et al. The C-509T promoter polymorphism of the transforming growth factor beta-1 gene is associated with levels of total and specific IgE in a Colombian population. Int Arch Allergy Immunol; 151(3): 237-46. [ Links ]
61. Rogers AJ et al. Assessing the Reproducibility of Asthma Candidate Gene Associations Using Genome-wide Data. Am J Respir Crit Care Med 2009. [ Links ]
62. Weidinger S et al. Genome-wide scan on total serum IgE levels identifies FCER1A as novel susceptibility locus. PLoS Genet 2008; 4(8): e1000166. [ Links ]
63. Rafaels NM et al. Validation of Previously Reported Genetic Associations for Asthma Using a Genome-Wide Association Approach among a Large Population of African Descent. Journal of Allergy and Clinical Immunology 2008; 121(2): S63. [ Links ]

