










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Salud Uninorte
Print version ISSN 0120-5552On-line version ISSN 2011-7531
Salud, Barranquilla vol.32 no.3 Barranquilla Sept./Dec. 2016
Fisiopatología molecular en la infección por Helicobacter pylori
Molecular pathophysiology in infection by Helicobacter pylori
Franklin Torres Jiménez1, Carlos Torres Bayona2
1Bacteriólogo, magíster en Inmunología, Universidad Libre, Facultad de Ciencias Exactas y Naturales. Barranquilla (Colombia).
2 Microbiólogo, magíster en Microbiología Molecular, Universidad Libre, Facultad de Ciencias Exactas y Naturales. Barranquilla (Colombia).
Correspondencia: Franklin Enrique Torres Jiménez. Cra 27 n°6-20, Km 7, antigua a vía Puerto Colombia. Primer piso. Celular: 3003042793, fijo: 3857341. franklintj654@hotmail.com, ftorres@unilireaq.edu.co.
Fecha de recepción: 25 de junio de 2016
Fecha de aceptación: 16 de septiembre de 2016
Resumen
La infección por Helicobacter pylori es el proceso de colonización bacteriana más común a nivel mundial. En 1994 la Agencia Internacional para la Investigación en Cáncer (IARC) catalogó al Helicobacter Pylori como un carcinógeno tipo I. La infección por esta bacteria incrementa el riesgo de padecer cáncer gástrico hasta 6 veces más que los individuos que no presentan la infección. Colombia tiene una prevalencia de infección por Helicobacter pylori del 86 % en adultos mayores de 20 años y del 80% en niños mayores de 8 años. La infección persistente y recidivante de la mucosa gástrica puede generar una respuesta inflamatoria, o procesos silentes, que en el peor de los casos puede progresar a un adenocarcinoma gástrico tipo difuso o intestinal. Existen múltiples factores que influyen en las consecuencias clínicas de la infección por Helicobacter pylori: en mención: predisposición genética del huésped, inmunorregulación genética del paciente infectado, factores ambientales y heterogeneidad de los factores de virulencia de las diversas cepas. Por lo anterior se infiere que las patologías gástricas, en especial la gastritis y el adenocarcinoma, son padecimientos de etiología multifactorial que pueden originarse por diversas causas o condiciones. En esta revisión se analizarán los principales aspectos relacionados con la fisiopatología molecular en la infección por Helicobacter pylori, en particular tópicos correspondientes a los factores de virulencia.
Palabras clave: infección, Helicobacter pylori, virulencia, inflamación, gastritis.
Abstract
Helicobacter pylori infection is the most common process worldwide bacterial colonization. In 1994 the International Agency for Research on Cancer (IARC) categorized the Helicobacter pylori as a type I carcinogen. The infection by this bacterium increases the risk of gastric cancer to 6 times more than individuals who do not have the infection. Colombia has a prevalence of Helicobacter pylori infection of 86% in adults aged 20 years and 80% in children over 8 years. Persistent infection and recurrent gastric mucosa can generate an inflammatory response, or silent processes, which in, the worst of the cases, can progress to gastric adenocarcinoma diffuse type or intestinal. There are multiple factors that influence the clinical consequences of Helicobacter pylori infection, some of them: host genetic predisposition, genetic immunoregulation of infected patients, environmental factors and heterogeneity of the virulence factors of the various strains. Therefore, we can say that gastric pathologies, especially gastritis and adenocarcinoma, are conditions of multifactorial etiology that may result from various causes or conditions. In this review, we analyze the main aspects related to the molecular pathophysiology in Helicobacter pylori infection, with emphasis on topics related to virulence factors.
Keywords: infection, Helicobacter pylori, virulence factors, inflammation, gastritis.
INTRODUCCIÓN
La infección por Helicobacter pylori es el proceso de colonización bacteriana más común a nivel mundial (1). El Centro para el Control y Prevención de Enfermedades calculó que casi dos tercios de la población mundial albergan la bacteria, y los índices de infección son mucho más elevados en los países en vía de desarrollo que en las naciones desarrolladas (2). En 1994, la Agencia Internacional para la Investigación en Cáncer (IARC) catalogó al H. Pylori como un carcinógeno tipo I en humanos, es decir, un carcinógeno definitivo (3). En la actualidad, la colonización del estómago por H. pylori se considera un factor de riesgo para el desarrollo de úlcera péptica, gastritis crónica, adenocarcinoma gástrico, linfoma gástrico de tejido linfoide asociado a mucosa (Maltoma), entre otras gastropatías (4).
Sin embargo, se ha demostrado que la infección por H. Pylori es necesaria, mas no suficiente, para el desarrollo de lesiones a nivel de la mucosa gástrica (5). Durante el proceso de colonización, infección y defensa se desencadenan eventos celulares y moleculares que pueden conllevar a la injuria de la mucosa gástrica; lo cual hace pensar que el proceso inflamatorio no solamente depende del H. pylori per se sino de algunas condiciones individuales de cada huésped infectado. Los mecanismos moleculares no están muy bien definidos, pero en la actualidad son motivo de investigación.
Otro aspecto relevante en la fisiopat ología de la infección por H. pylori es la diversidad o heterogeneidad de las cepas. Hoy en día se sabe que no todas las cepas de H. pylori son patógenas, y que la presencia de ciertos factores de virulencia marca la diferencia entre las que se consideran microbiota de la mucosa gástrica y las que están asociadas con gastritis crónica y adenocarcinoma gástrico (6, 7). También existe la evidencia de cepas altamente resistentes a la antibioticoterapia empleada en los esquemas terapéuticos usados en gastroenterología, que convierten este proceso infeccioso en un verdadero problema prioritario de salud pública a nivel nacional e internacional.
En esta revisión se analizarán los principales aspectos relacionados con la fisiopatología molecular en la infección por H. pylori, en particular tópicos correspondientes a los factores de virulencia.
PATOGENIA EN LA INFECCIÓN POR HELICOBACTER PYLORI
Helicobacter pylori ingresa por la boca, desciende al tubo digestivo y a través de sus flagelos se transporta hasta la superficie de la capa de mucus que recubre las células epiteliales de la mucosa gástrica del fundus y antro pilórico preferiblemente (8). H. pylori posee adhesinas que favorecen su adherencia a las células foveolares superficiales (9). La colonización se facilita por la inhibición de la producción de ácido clorhídrico (HCl) y la neutralización de este por el amonio producido por la acción de la ureasa bacteriana (10).
H. pylori provoca citotoxicidad a nivel de la mucosa gástrica debido a un sistema de secreción tipo IV, codificado por genes ubicados en una región genómica de 37 kb denominada "Isla de patogenicidad CagA o Cag-PAI", que facilita la inyección de proteínas con actividad citopática como CagA y Vac A, respectivamente (11).
H. pylori posee fosfolipasas que hidrolizan las membranas celulares, lo cual conlleva a la liberación de lisolecitinas, las cuales constituyen un factor ulcerogénico (12). También posee lipopolisacáridos (LPS), peptidoglucanos, tetrapéptidos, entre otros PAMPs (Patrones Moleculares Asociados a Patógenos) que estimulan a una gran variedad de receptores extra- e intracelulares como el Nod 1, los cuales ejercen un importante efecto quimiotáctico sobre los eosinófilos y neutrófilos, y facilitan su reclutamiento y proliferación (13). Estas células al activarse provocan la liberación de citoquinas, lo cual desencadena una respuesta inflamatoria amplificante, la cual lesiona aun más la mucosa mediante la liberación de mediadores inflamatorios (14).
FACTORES DE VIRULENCIA QUE CONTRIBUYEN A LA COLONIZACIÓN DE LA MUCOSA GÁSTRICA
En la Tabla 1 se listan los principales factores de virulencia que contribuyen al proceso de colonización por H. pylori. A continuación se detallan algunos aspectos relevantes de cada uno de ellos.

Ureasa: Hidroliza la urea [CO(NH2)2] en amonio (NH4+) y gas carbónico (CO2). Proporciona un pH neutro alrededor del microorganismo, que le permite evadir las propiedades bactericidas del ácido clorhídrico (HCl). Los iones amonio inhiben los trasportadores gástricos de bicarbonato, con lo cual se impide la alcalinización de la mucosa gástrica (15).El amonio producido aumenta el pH, elevándolo hasta 6 o 7 en su entorno. De este modo puede alcanzar la superficie de las células de la mucosa, donde el pH es prácticamente neutro.El NH4+ produce una serie de daños que afectan a la microcirculación y a las células epiteliales superficiales y origina una necrotización del tejido.
Sistemas antioxidantes: Durante el proceso de colonización H. pylori promueve una fuerte respuesta inflamatoria mediada por neutrófilos y macrófagos, que generan una cantidad de metabolitos reactivos del oxígeno. H. pylori cuenta con mecanismos para la detoxificación de estos metabolitos, así como para la reparación de los daños sufridos que favorecen su supervivencia en el tejido inflamado (16).
Entre los sistemas enzimáticos de detoxificación están: la enzima superóxido dismutasa (cataliza la transformación del superóxido en peróxido de hidrogeno), la catalasa o peroxidasa (cataliza la descomposición del peróxido de hidrógeno (H2O2), en agua y oxigeno gaseoso). También facilita la evasión de la fagocitosis realizada por macrófagos ubicados en el epitelio superficial (17); las peroxirredoxinas (catalizan la reducción de peróxido de hidrógeno, peroxinitrito y otros hidroperóxidos orgánicos a sus correspondientes alcoholes), la flavoproteína MdaB, una NADPH quinona reductasa, que H. pylori expresa cuando debe compensar la pérdida de los principales componentes antioxidantes.
La proteína NAP (Neutrophil activating protein), codificada por el gen napA, tiene función de bacterioferritina para captar los iones ferrosos libres intracelulares que pueden dañar el DNA de H. pylori y protege a este del estrés oxidativo. Puede actuar como adhesina cuando se secreta o se expresa en la superficie bacteriana, pues tiene afinidad por las ceramidas presentes en las membranas plasmáticas celulares y por el grupo sanguíneo Lewis (18).
Flagelos: Facilitan la penetración y la adherencia en el epitelio superficial de la mucosa gástrica (19). H. pylori posee alrededor de 2 a 6 flagelos monopolares. Cada flagelo está compuesto por dos flagelinas, FlaA y FlaB. FlaB se localiza en la base del flagelo, mientras que la más abundante, FlaA, se encuentra en el exterior.
Adhesinas: H. pylori se une a las células del huésped de forma específica mediante un elevado número de adhesinas. A continuación se mencionan las adhesinas involucradas en la fisiopatología de la infección por H. pylori:
HpaA (Helicobacter pylori adhesin A): es una de las principales proteínas de la membrana externa. HpaA media la unión a glicoconjugados con ácido siálido (N-acetil-neuraminil-lactosa) presentes en la superficie de las células epiteliales gástricas y neutrófilos (20).
BabA (Blood group antigen-binding adhesion): codificada por los genes babA1 y babA2, aunque solo el gen babA2 es funcionalmente activo. BabA se une al antígeno del grupo sanguíneo B y al antígeno Lewis ubicado en la mucosa gástrica (21). Esta unión promueve una respuesta inmune no específica con producción de autoanticuerpos dirigidos a las células productoras de HCL, lo cual contribuye a la gastritis crónica y a la pérdida de células parietales. Además, la adherencia mediada por BabA participa en la distribución de los factores de virulencia que dañan al tejido del hospedador, pudiendo llevar al desarrollo de ulcera y cáncer gástrico (22).
SabA (Sialic acid-binding adhesion): proteína de adhesión al ácido siálico, se une a los receptores con el ácido siálico de los neutrófilos y origina la activación de su respuesta oxidativa (23).
OipA (Outer inflammatory protein): proteína inflamatoria externa; todas las cepas poseen el gen que codifica para esta adhesina, pero solo algunas la expresan (24). Su expresión está asociada a una mayor producción de IL-8 y otras citoquinas proinflamatorias, aunque no se sabe cuál es su contribución real en la inflamación gástrica, puesto que suele estar asociada a las cepas cagA+. OipA también está asociada con el desarrollo de úlcera duodenal y gastritis (25).
FACTORES DE VIRULENCIA QUE CONTRIBUYEN AL DAÑO DE LA MUCOSA GÁSTRICA
En la actualidad son los factores de virulencia que más se estudian a nivel mundial, debido a su estrecha relación con los procesos de carcinogénesis y citotoxicidad. En la tabla 2 se enumeran los principales factores de virulencia asociados al daño de la mucosa gástrica. A continuación se desglosan los procesos moleculares más relevantes de cada uno de ellos.
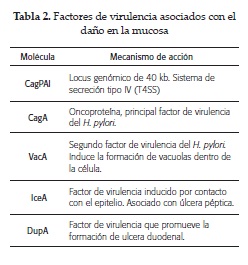
Isla de patogenicidad CagA (CagPAI): locus genómico de 40 kb, que contiene27 a 31 genes codificantes para proteínas que participan en el sistema de secreción tipo IV (T4SS); considerado como una "Jeringa molecular" (26). Los genes de la CagPAI no se expresan constitutivamente, sino que responden a señales ambientales. Están regulados por mecanismos complejos que pueden activar o inhibir, dependiendo de condiciones microambientales como el nivel de oxígeno, la osmoralidad, la fase de crecimiento bacteriano, el pH, presencia o no de ácidos grasos volátiles de cadena corta, entre otras condiciones.
La CagPAI codifica un sistema de secreción de proteínas tipo IV que inyecta CagA y peptidoglucanos en las células epiteliales del hospedador. La translocación de cagA depende de la presencia de un canal de urea protón dependiente UreI. Ante un descenso de pH, cagA se mueve del centro a la porción periférica del citoplasma. La proteína inyectada interactúa con diversas moléculas de la célula hospedadora. La proteína CagA translocada aumenta la producción de IL-8. Así mismo, los productos de la CagPAI están asociados con un aumento de la producción de otras citoquinas como la IL-1β, TNF-α y la molécula NF-κβ (27).
CagA (Citotoxina asociada al gen del antígeno A): se considera el principal factor de virulencia del H. pylori (28). Cuando esta proteína ingresa a la célula es fosforilada por la acción de tirosinas kinasas intracelulares, lo cual genera alteraciones en la traducción de señales, que conducen a cambios proliferativos e inflamatorios asociados con el desarrollo de úlcera y cáncer. No todas las cepas de H. pylori presentan este factor de virulencia. La diana molecular de CagA es una fosfatasa SHP-2 (proteína tyrosine phospatase) (29).
En el gen CagA se han encontrado mutaciones y polimorfismos que están relacionados con la carcinogénesis gástrica (29). La propia activación de SHP-2 por CagA puede contribuir a la proliferación celular excesiva; además, los cambios que se producen en la expresión génica en las células epiteliales tras la infección por H. pylori suelen ser dependientes de este sistema de secreción codificado por la CagPAI.
CagA puede variar de tamaño entre las distintas cepas de H. pylori. Esta variación proviene de la presencia de un número de repeticiones de una secuencia aminoacídica del extremo C-terminal y puede influir en la patogenicidad de las distintas cepas cagA+, debido a que la variación en el número de sitios de fosforilación implica una distinta efectividad en su unión a SHP-2, y por tanto una distinta activación (30).
La variabilidad en la región C-terminal de la proteína cagA consiste en el número de repeticiones de la secuencia Glu-Pro-Ile-Tyr-Ala, llamado "Motivo EPIYA" ; algunas cepas cagA+ carecen de él. La secuenciación de este motivo ha revelado que la región que flanquea esta secuencia de aminoácidos presenta cuatro segmentos distintos, conocidos como EPIYA-A, EPIYA-B, EPIYA-C y EPIYA-D (31).
EPIYA-A y B están presentes en casi todas las cepas cagA+. EPIYA-C es característico de cepas de H. pylori cagA+ presentes en Europa, Norteamérica y Australia, por lo cual es llamado "cagA Occidental" (32). EPIYA-D es específico de cepas circulantes en países de Asia Oriental, como Korea, Mongolia, Japón y China; por lo tanto se conoce como "cagA de Asia Oriental" (33). Los estudios sobre la proteómica de la oncoproteína cagA han revelado que el motivo EPIYA puede repetirse hasta tres veces, sin embargo, el número puede variar desde uno hasta siete repeticiones. La cepa que contiene el segmento EPIYA-D tiene un mayor potencial oncogénico que la cepa EPIYA-C. Sin embargo, se ha demostrado que cepas que contienen el genotipo Occidental presentan gran poder de fosforilación y oncogénico dependiendo del número de repeticiones del motivo EPIYA-C (31-33). Por lo anterior se ha postulado que las distintas isoformas de la oncoproteína cagA pueden influir en las diferentes tasas de cáncer gástrico observadas en países orientales y occidentales.
VacA (Citotoxina de vacuolización A): la proteína VacA induce la formación de vacuolas dentro de la célula, también impide la fagocitosis, altera la presentación antigénica y promueve la apoptosis de la célula epitelial (34). Está asociada al desarrollo de úlcera y adenocarcinoma gástrico. Distinto de lo que sucede con el gen cagA, todas las cepas de H. pylori son vacA positivas, aunque solo casi el 50 % de estas expresan la proteína.
VacA posee una estructura hexamérica y se ensambla en la bicapa lipídica celular del hospedador formando un canal selectivo de aniones. Produce un gradiente de pH que atrae sustancias alcalinas al interior haciendo que se capte agua por ósmosis, lo cual origina una vacuolización alrededor del núcleo y más tarde el estallido y muerte celular (35).
El gen vacA es altamente polimórfico, y presenta diversidad en algunas regiones de la estructura molecular (36). Por ejemplo: a nivel de la secuencia señal 5´ terminal se han identificado los alelos s1a, s1b, s1c y s2. En la región media los alelos m1 y m2, y en la región intermedia los alelos i1 e i2. Esta diversidad genética causa variación en la actividad citotóxica de la proteína VacA.
Se ha demostrado que las cepas s1m1 y s1m2 son altamente toxigénicas, mientras que las cepas con la combinación s2m2 producen muy poca citotoxicidad. Las cepas que contienen la isoforma i1 están asociadas con un alto riesgo de desarrollar adenocarcinoma gástrico (36). La cepa VacA s1a predomina en el norte de Europa, la s1b en la península ibérica y Latinoamérica, mientras que la cepa s1c se encuentra en el suroriente de Asia. En Colombia se ha demostrado la presencia de la cepa que contiene los alelos s1m1, al igual que en Japón, siendo ambos países regiones con una alta tasa de cáncer gástrico (37).
VacA puede promover la apoptosis de forma independiente a la vacuolización, pues induce la liberación de citocromo c mitocondrial a través de la activación de proteínas proapoptóticas Bax y Bak, al igual que la activación del receptor Fas/CD95 (38).
La presencia de vacA puede inducir la expresión del factor de crecimiento vascular endotelial (VEGF) y provocar el desarrollo de procesos tumorigénicos. El VEGF está implicado en la neoangiogénesis y se encuentra sobreexpresado en carcinomas humanos que contienen cepas vacA+ (39). Además, VacA amplifica la respuesta inflamatoria de la mucosa gástrica aumentando la expresión de ciclooxigenasa 2 en las células T, neutrófilos y macrófagos, que a su vez pueden activar la producción del factor de crecimiento vascular endotelial y provocar el desarrollo de procesos tumorigénicos.
Aunque no están perfectamente definidos los mecanismos por los que la respuesta inmune inducida por H. pylori contribuye a la carcinogénesis gástrica, la sobreexpresión de COX-2 y VEGF, sumado a la activación de NF-κβ y citoquinas proinflamatorias, originan alteraciones morfológicas que llevan al desarrollo de gastritis atrófica y metaplasia gastrointestinal (40).
VacA también está implicada en la alteración de las funciones mediadas por integrinas al interactuar con la fibronectina y la modulación de la respuesta inmunitaria de granulocitos, monocitos, células B y T, ya que inhibe la presentación de antígenos y la proliferación de células T. Por otro lado, interrumpe la maduración de los fagosomas en los macrófagos, por lo que la bacteria sobrevive dentro de los mismos (41).
Otro aspecto relevante de VacA es que inhibe la activación del factor de transcripción NF-AT (Factor Nuclear Activador de la Transcripción), una molécula importante para la expresión de genes implicados en la expansión de linfocitos T activados por antígenos bacterianos. Asegurando así la evasión del H. pylori por parte de la respuesta inmune celular adaptativa (42).
4.4 IceA (Induced by contact with epithelium): factor de virulencia inducido por contacto con el epitelio, es codificado por el gen IceA, el cual posee dos variantes, el iceA1 e iceA2. La variante iceA1 está asociada con el desarrollo de úlcera péptica, mientras que la variante iceA2 todavía no tiene asociación definida (43).
DupA (Duodenal ulcer-promoting gene): factor de virulencia que promueve la formación de ulcera duodenal, tiene un alto potencial patogénico (44).
OTROS FACTORES DE VIRULENCIA
LPS (Lipopolisacáridos): el LPS de H. pylori, como el de otras especies bacterianas, presenta una estructura de tres dominios principales: la capa polisacárida externa o cadena específica O, el núcleo oligosacárido y el lípido A (45). Son patrones moleculares presentes en la membrana de la bacteria, muy común en bacterias Gram negativas. Son considerados potentes endotoxinas promotoras de respuesta inflamatoria. Son reconocidos por los Toll Like Receptor (TLR) tipo 4 ubicados en la membrana de la célula epitelial de la mucosa gástrica, importantes en la respuesta inmune innata (46). La estructura química del LPS de H. pylori, la cadena específica O, puede mimetizar los antígenos del grupo sanguíneo Lewis.
Tip α(TNF-α inducing protein): La proteína Tip α tiene una potente actividad carcinogénica a través de la inducción de TNF-α y la activación del NF-kβ, lo cual favorece a la inflamación y al cáncer (47).
FACTORES DE VIRULENCIA: EVIDENCIA CLÍNICA Y EPIDEMIOLOGÍA MOLECULAR
La gastritis aguda por H. pylori cursa con epigastralgía, náuseas y vómitos (dispepsia no ulcerosa); mientras que la gastritis crónica se caracteriza por infiltración inflamatoria linfocítica, con folículos linfoides y un grado variable de actividad celular (48,49).
En Colombia se han publicado algunos estudios de tipificación molecular de cepas de H. pylori halladas en biopsias gástricas. Molina y cols. (50) analizaron 60 muestras de biopsias gástricas de pacientes que manifestaban dispepsia. Se logró identificar, por medio de pruebas de biología molecular, el factor de virulencia CagA en 16 pacientes (43,24 %). De los cuales 13 presentaban gastritis no atrófica, 3 gastritis atrófica multifocal, 1 displasia y metaplasia intestinal y 1 carcinoma gástrico. Se observó que CagA está presente en las lesiones más severas de la gastritis, metaplasia intestinal + displasia y carcinoma gástrico.
Trujillo y cols. (51) genotipificaron los factores de virulencia vacA y cagA en individuos de dos regiones de Colombia con riesgo opuesto de cáncer gástrico. Se analizaron 401 biopsias del antro gástrico provenientes de individuos con diagnóstico de gastritis no atrófica, gastritis atrófica y metaplasia intestinal; 256 se obtuvieron en la zona de alto riesgo (Tunja y Bogotá) y 145 en la zona de bajo riesgo (Barranquilla, Santa Marta y Cartagena). No se observó diferencia en la frecuencia de infección por H. pylori entre las dos zonas (77,3 vs. 77,9 %, p=≥ 0,05). La presencia de cagA fue mayor en la zona de bajo riesgo (77,9 vs. 69,2 %, p=ns). El alelo vacA s1 fue más prevalente en la zona de bajo riesgo (61,8 vs. 72,0 %, p=ns). El alelo vacA m1 presentó mayor prevalencia en la zona de alto riesgo (57,2 vs. 42,8 %, p=ns). La combinación cagA+ s1m1 también fue más frecuente en la zona de bajo riesgo (48,9 vs. 38,9 %, p=ns).
Los resultados observados anteriormente contrastan con los resultados obtenidos por Bravo y cols. (52), los cuales genotipificaron los factores de virulencia CagA y vacA en 241 muestras obtenidas de biopsias gástricas provenientes de áreas de alto riesgo de cáncer gástrico (Pasto) y 93 muestras provenientes de áreas de bajo riesgo (Tumaco). Las frecuencias relativas más altas de los genotipos cagA, vacA s1 y vacA m1 fueron estadísticamente significativas en pacientes provenientes de la población con alto riesgo de cáncer (Pasto) en comparación con la población de bajo riesgo (Tumaco). Concluyen los autores que estos resultados podrían explicar el "enigma africano", en donde hay poblaciones con una gran prevalencia de infección por H. pylori pero una baja prevalencia de cáncer gástrico.
Recientemente Kodaman y cols. (53) realizaron un estudio en Túquerres, zona montañosa de alto riesgo al sur de Colombia, y en Tumaco, población de la costa pacífica con bajo riesgo de cáncer gástrico; reportaron que en los aislamientos de H. pylori de la zona de bajo riesgo predominó una cepa de ancestro africano, mientras que en la zona de alto riesgo predominó una de ancestro europeo. Por otro lado, se observó el predominio del ancestro africano en los individuos infectados de la costa y del ancestro amerindio en los individuos infectados de la zona montañosa. La hipótesis de estos autores se basa en que la coevolución entre las cepas de H. pylori y el huésped son los responsable del riesgo de manifestar patologías gástricas.
Algunos autores respaldan la idea de que la tipificación genética de cepas de H. pylori podría ser superior que los marcadores genéticos humanos para distinguir poblaciones relacionadas geográficamente. A través de la técnica de análisis de tipificación de secuencia multilocus (MLST: multilocus sequence typing) de genes estructurales (housekeeping) han arrojado resultados robustos y consistentes para examinar ancestría y evolución de poblaciones de H. pylori (54).
Falush y cols. (54), utilizando la técnica MLST, analizaron los genes estructurales atpA, efp, mutY, ppa, trpC, ureI, yphC, más el vacA, de 370 cepas aisladas de 27 poblaciones humanas distintas. Encontraron 4 grupos principales de poblaciones de H. pylori con distinta distribución geográfica, las cuales fueron identificadas como hpEurope, hpAfrica1, hpAfrica2 y hpEastAsia. Un análisis más detallado dividió el grupo hpEastAsia en tres subpoblaciones: hspAmerind, hspEAsia and hspMaori. El grupo hpAfrica1 fue dividido en hspWAfrica and hspSAfrica, respectivamente. Estudios más recientes han identificado más grupos hpNEAfrica, hpAsia2 y hpSahul. En la tabla 3 se resume la distribución filogeográfica del H. pylori.

Estos hallazgos son de vital importancia, ya que la presencia de cepas virulentas son necesarias, mas no suficientes, a la hora de iniciar un proceso de carcinogénesis; hay que tener en cuenta el prototipo de la cepa y determinar su ancestría poblacional. Por lo tanto, queda establecido que en la población de las montañas andinas colombianas predomina el prototipo europeo (hpEurope, en un 100%), mientras que en la costa pacífica colombiana predomina el prototipo africano (hpAfrica 1, en un 65,5 %. El 34,5 % restante son hpEurope). Cabe resaltar que el prototipo hpAfrica 1 posee dos subpoblaciones: hspSAfrica y hspWAfrica. La distribución de estos en la costa pacífica colombiana fue de 3 y 16, con un total de 19 cepas hpAfrica 1.
En este mismo estudio se analizó la ancestría humana en las dos poblaciones, junto con la severidad de las lesiones en la mucosa gástrica. Se evidenció una gran diferencia entre las dos regiones. En la población de la costa pacífica se observó una ancestría africana del 58 %, mientras que en la población montañosa se evidenció una ancestría amerindia, en un 67 %. En cuanto a los resultados de histopatología, las lesiones observadas en la mucosa de los pacientes con ascendencia africana eran relativamente benignas; contrario a las lesiones observadas en los amerindios, que presentaban resultados más deletéreos (lesiones más severas).
Los datos observados en las regiones afrodescendientes de Colombia son coincidentes con lo observado por Holcombe y cols. en 1992 (55), investigación en la que se llamó "Enigma Africano" a lo observado en varias poblaciones africanas, pues presentan un 100 % de prevalencia de infección por H. pylori, pero baja tasa de adenocarcinoma gástrico (55). Dicha investigación deja en evidencia que otros factores diferentes de la infección podrían jugar un papel protagónico en la etiopatogenia de la gastritis y sus complicaciones. También deja ver cómo algunas propiedades o condiciones intrínsecas del huésped influyen en la tolerancia ante la infección por H. pylori.
Entre las investigaciones futuras de nuestro grupo de investigación se considera estudiar la ancestría de las cepas de H. pylori aisladas en Barranquilla y compararlas con las reportadas en el territorio colombiano. Nuestro interés se fundamenta en que gran parte de la población de la costa pacífica colombiana es mulata, al igual que la población de la región Caribe. Por lo tanto, esperaríamos encontrar resultados coincidentes o reproducibles que sigan enriqueciendo los aspectos moleculares asociados a la infección por H. pylori.
Conflicto de intereses: ninguno.
Financiación: Universidad Libre Seccional Barranquilla.
REFERENCIAS
1. Bucley MJM, O ́Morain CA. Helicobacter pylori biology-discovery. Br Med Bull 1998; 54: 7-16. [ Links ]
2. American College of Gastroenterology guideline on the management of Helicobacter pylori infection. Bethesda, Md.: American College of Gastroenterology.http://www.acg.gi.org/physicians/guidelines/ManagementofHpylori.pdf. Acceso 10 de Junio de 2013. [ Links ]
3. Schistosomes, liver flukes and Helicobacter pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, 7-14 June 1994. IARC Monogr Eval Carcinog Risks Hum 1994; 61: 1-241. [ Links ]
4. Amieva MR, El-Omar EM. Host-bacterial interactions in Helicobacter pylori infection. Gastroenterology 2008; 134:306-323. [ Links ]
5. Blaser MJ, Berg DE. Helicobacter pylori genetic diversity and risk of human disease. J Clin Invest. 2001; 107:767-73. [ Links ]
6. Mager DL. Bacteria and cancer: ¿cause, coincidence or cure? A review. J Translat Med 2006; 4:14. [ Links ]
7. International Agency for Research on Cancer. Globocan 2012. Fecha de consulta: 12 de junio de 2015. Disponible en: http://globocan.iarc.fr/Pages/fact_sheets_cancer.aspx. [ Links ]
8. Bai H, Li Q, Liu X, Li Y. Characteristics and Interactions of Helicobacter pylori and H. pylori-Infected Human Gastroduodenal Epithelium in Peptic Ulcer: A Transmission Electron Microscopy Study. Dig Dis Sci 2009. [ Links ]
9. Mahdavi J et al. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science 2002; 297: 573-578. [ Links ]
10. Dunn, B. E et al. Localization of Helicobacter pylori urease and heat shock protein in human gastric biopsies. Infect. Immun 1997; 65:1181-1188. [ Links ]
11. Atherton JC, Cao P, Peek RM Jr, Tummuru MK, Blaser MJ, Cover TL. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J Biol Chem 1995; 270:17771-7. [ Links ]
12. Ramis IB et al. Molecular Basis of pathogenicity in Helicobacter pylori clinical isolates. J Clin Microbiol 2010; 48: 3776-3778. [ Links ]
13. Masumoto J et al. Nod1 acts as an intracellular receptor to stimulate chemokine production and neutrophil recruitment in vivo. J Exp Med 2006; 203: 203-213. [ Links ]
14. Fritz JH et al. Nod1-mediated innate immune recognition of peptidoglycan contributes to the onset of adaptive immunity. Immunity 2007; 26: 445-459. [ Links ]
15. Hatakeyama M. Helicobacter pylori and gastric carcinogenesis. J Gastroenterol 2009; 44: 239-248. [ Links ]
16. De Falco M, Lucariello A, Iaquito S, Esposito V, Guerra G, De Luca A. Molecular mechanisms of Helicobacter pylori patogénesis. J Cell Physiol 2015; 29:1702-7. [ Links ]
17. Portal-Celhay C, Perez-Perez GI. Immune responses to Helicobacter pylori colonization: mechanisms and clinical outcomes. Clin Sci (Lond) 2006; 110: 305-314. [ Links ]
18. Long M, Luo J, Li Y, Zeng FY, Li M. Detection and evaluation of antibodies against neutrophil-activating protein of Helicobacter pylori in patients with gastric cancer. World J Gastroenterol 2009; 15: 2381-8. [ Links ]
19. Yokota S, Ohnishi T, Muroi M, Tanamoto K, Fujii N, Amano K. Highly-purified Helicobacter pylori LPS preparations induce weak inflammatory reactions and utilize Toll-like receptor 2 complex but not Toll-like receptor 4 complex. FEMS Immunol Med Microbiol 2007; 51: 140-148. [ Links ]
20. Nvström J, Svennerholm AM. Oral immunization with HpaA affords therapeutic protective immunity against H. pylori that is reflected by specific mucosal immune responses. Vaccine 2007: 25; 2591-8. [ Links ]
21. Hennig EE, Mernaugh R, Edl J, Cao P, Cover TL. Heterogeneity among Helicobacter pylori Strains in expression of outer membrane protein BabA. Infect Immunol 2004; 72: 3429-3435. [ Links ]
22. Rad R et al. The Helicobacter pylori blood group antigen-binding adhesin facilites bacterial colonization and augment a nonspecific immune response. J Immunol 2002; 168: 3033-3041. [ Links ]
23. Unemo M, Aspholm-Hurtig M, Ilver D, Bergstrom J, Boren T, Danielsson D, Teneberg S. The sialic acid binding SabA adhesion of Helicobacter pylori essential for nonopsonic activation of human neutrophils. J Bil Chem 2005; 280:15390-15397. [ Links ]
24. Yamaoka Y, Kikuchi S, el-Zimaity HM, Gutierrez O, Osato MS, Graham DY. Importance of Helicobacter pylori OipA in clinical presentation, gastric inflammation, and mucosal interleukin 8 production. Gastroenterology 2002; 123: 414-424. [ Links ]
25. Yamaoka Y, Kwon DH, Graham DY. A M(r) 34,000 proinflammatory outer membrane protein (oipA) of Helicobacter pylori. Proc Natl Acad Sci USA 2000; 97: 7533-7538. [ Links ]
26. Backert S et al. Translocation of the Helicobacter pylori CagA protein in gastric epithelial cells by a type IV secretion apparatus. Cell Microbiol 2000; 2: 155-164. [ Links ]
27. Kumar Pachathundikandi S, Brandt S, Madassery J, Backert S. Induction of TLR-2 and TLR-5 expression by Helicobacter pylori switches cagPAI-dependent signalling leading to the secretion of IL-8 and TNF-α. PLoS One 2011; 6: e19614. [ Links ]
28. Backert S et al. Translocation of the Helicobacter pylori CagA protein in gastric epithelial cells by a type IV secretion apparatus. Cell Microbiol 2000; 2: 155-164. [ Links ]
29. Higashi H, et al. Helicobacter pylori CagA induces Ras-independent morphogenetic response through SHP-2 recruitment and activation. J Biol Chem 2004; 279: 17205-17216. [ Links ]
30. Odenbreit S, Plus J, Sedlmair B, Gerland E,Fisjer W, Haas R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 2000; 287: 1497-1500. [ Links ]
31. Panayotopoulou E, et al. Strategy to characterize the number and type of repeating EPIYA phosphorylation motifs in the carboxyl terminus of CagA protein in Helicobacter pylori clinical isolates. J Clin Microbiol 2006; 45: 488-95. [ Links ]
32. Hatakeyama M. Anthropological and clinical implications for the structural diversity of the Helicobacter pylori CagA oncoprotein. Cancer Sci 2011; 102:36-43. [ Links ]
33. Truong BX et al. Diverse characteristics of the CagA gene of Helicobacter pylori strains collected from patients from southern Vietnam with gastric cancer and peptic ulcer. J Clin Microbiol 2009; 47:4021-4028. [ Links ]
34. Basso D et al. Clinical relevance of Helicobacter pylori cagA and vacA gene polymorphisms. Gastroenterology 2008; 135: 91-99. [ Links ]
35. Yang JJ et al. Oncogenic CagA promotes gastric cancer risk via activating ERK signaling pathways: a nested case-control study. PLoS One 2011; 6: e21155. [ Links ]
36. Rhead JL et al. A new Helicobacter pylori vacuolating cytotoxin determinant, the intermediate region, is associated with gastric cancer. Gastroenterology 2007; 133: 926-936. [ Links ]
37. Trujillo E, Martínez T, Bravo MM. Genotipificación de los factores de virulencia vacA y cagA de Helicobacter pylori en individuos de dos regiones de Colombia con riesgo opuesto de cáncer gástrico. Biomédica 2014; 34:567-73. [ Links ]
38. Allen LA, Schlesinger LS, Kang B. Virulent strains of Helicobacter pylori demonstrate delayed phagocytosis and stimulate homotypic phagosome fusion in macrophages. J Exp Med 2000; 191: 115-128. [ Links ]
39. Blaser MJ et al. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res 1995; 55: 2111 - 15. [ Links ]
40. Ando T, Gots G, Ishiguro K, Malda O, Watanabe O, Omuya N. The interaction of host genetic factors and Helicobacter pylori infection. Inflammation Pharmachology 2007; 15: 10-14. [ Links ]
41. Wilson KT, Crabtree JE. Immunology of Helicobacter pylori: insights into the failure of the immune response and perspectives on vaccine studies. Gastroenterology 2007; 133:288-308. [ Links ]
42. Sundrud MS, Torres VJ, Unutmaz D, Cover TL. Inhibition of primary human T cell proliferation by Helicobacter pylori vacuolating toxin (VacA) is independent of VacA effects on IL-2 secretion. Proc Natl Acad Sci USA 2004; 101: 7727-7732. [ Links ]
43. Peek RM et al. Adherence to gastric epitelial cells induces expression of a Helicobacter pylori gene, iceA, that is associated with clinical outcome. Proc Assoc Am Physicians 1998; 110: 531-544. [ Links ]
44. Shiota S, Matsunari O, Watada M, Hanada K, Yamaoka Y. Systematic review and meta-analysis: the relationship between the Helicobacter pylori dupA gene and clinical outcomes. Gut Pathogens 2010, 2:13. [ Links ]
45. Matsuura M. Structural Modifications of Bacterial Lipopolysaccharide that Facilitate Gram-Negative Bacteria Evasion of Host Innate Immunity. Front Immunol 2013; 4: 109. [ Links ]
46. Yokota S, Ohnishi T, Muroi M, Tanamoto K, Fujii N, Amano K. Highly-purified Helicobacter pylori LPS preparations induce weak inflammatory reactions and utilize Toll-like receptor 2 complex but not Toll-like receptor 4 complex. FEMS Immunol Med Microbiol 2007; 51: 140-148. [ Links ]
47. Suganuma M, Kuzuhara T, Yamaguchi K, Fujiki H. Carcinogenic Role of Tumor Necrosis Factor-α Inducing Protein of Helicobacter pylori in Human Stomach. BME 2006; 31:1 - 8. [ Links ]
48. Bornschein J, Kandulski A, Selgrad M, Malfertheiner P. From gastric inflammation to gastric cancer. Dig Dis 2010;28: 609-614. [ Links ]
49. Di Lorenzo. Subcomittee on chronic abdominal pain. Chronic Abdominal pain in children. Pediatrics 2005; 115: 370-381. [ Links ]
50. Molina Delgado AP, Jaramillo Henao CA, Delgado Perafán MP, Bohórquez Lozano ME y Ámezquita Torres A. Detección y genotipificación de Helicobacter pylori sobre la base de los genes ADNr 16S y el gen asociado a citotoxina (cagA) y posible asociación con enfermedades gastrointestinales. Rev cubana med trop 2008;60(2):105-10. [ Links ]
51. Trujillo E, Martínez T, Bravo MM. Genotipificación de los factores de virulencia vacA y cagA de Helicobacter pylori en individuos de dos regiones de Colombia con riesgo opuesto de cáncer gástrico. Biomédica 2014; 34:567-73. [ Links ]
52. Bravo LE, van Doom LJ, Realpe JL, Correa P. Virulence-associated genotypes of Helicobacter pylori: do they explain the African enigma? Am J Gastroenterol 2002 Nov;97(11):2839-42. [ Links ]
53. Kodaman N et al. Human and Helicobacter pylori coevolution shapes the risk of gastric disease. Proc Natl Acad Sci USA 2014; 111:1455-60. [ Links ]
54. Falush D, Wirth T, Linz B et al. Traces of human migrations in Helicobacter pylori populations. Science 2003; 299:1582-1585. [ Links ]
55. Holcombe C. Helicobacter pylori: the African enigma. Gut 1992; 33: 429-431. [ Links ]

