










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Salud Uninorte
Print version ISSN 0120-5552On-line version ISSN 2011-7531
Salud, Barranquilla vol.32 no.3 Barranquilla Sept./Dec. 2016
Anemia de células falciformes: una revisión
Sickle Cell Anemia: A review
Alfonso J. Ayala Viloria1, Henry J. González Torres2, Gabriel J. David Tarud3
1 Médico, residente de tercer año de Esp. Pediatra. Universidad Simón Bolívar (Barranquilla, Colombia). hgonzalez11@unisimonbolivar.edu.co.
2 Biólogo, Esp Stat. Apl., MSc Biología (Genética). Universidad Simón Bolívar (Barranquilla, Colombia). alfonsoayala81653@gmail.com.
3 Médico, pediatra, hematólogo/oncólogo pediátrico. Clínica de la Costa (Barranquilla, Colombia). gabrieldavidtarud@gmail.com.
Correspondencia: Alfonso J. Ayala Viloria. Universidad Simón Bolívar (Barranquilla, Colombia).
Fecha de recepción: 7 de julio de 2016
Fecha de aceptación: 13 de septiembre de 2016
Resumen
La anemia hemolítica más frecuente en la población mundial es la anemia de células falciformes (ACF), con una incidencia de 1/600 recién nacidos en Estados Unidos y en algunas regiones de España con incidencia de 1/5000 neonatos; en Colombia no hay registros respecto a la incidencia y prevalencia.
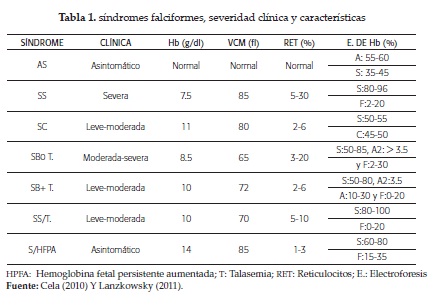
La transmisión de la ACF es autosómica dominante. Los homocigotos (SS) no sintetizan Hb A y poseen eritrocitos con un 90 % de Hb S. El portador o heterocigoto (AS) tiene hematíes con Hb A mayor que 50 % y Hb S de 20 - 40 % y son usualmente asintomáticos.
La Hb S se debe a una mutación en el gen de la cadena beta de globina, lo cual conlleva a la polimerización de la Hb en condiciones de baja oxigenación, lo cual origina un cambio en la morfología del eritrocito que adquiere la forma falciforme.
La sintomatología es secundaria a la anemia hemolítica crónica, la vaso-oclusión en los diferentes órganos y la asplenia funcional, la cual predispone a la infección. Otras manifestaciones asociadas son el secuestro esplénico, la aplasia eritroide y las complicaciones órgano - especificas, que disminuyen la calidad de vida y predisponen a mayor mortalidad.
Su manejo debe realizarse en centros de referencia donde haya un manejo integral, incluyendo el recurso humano y físico, ya que el manejo inadecuado y sus complicaciones disminuyen la sobrevida, la cual no es superior a los 45 años según reportes.
Palabras clave: anemia hemolítica, síndrome falciforme, muerte en edad pediátrica.
Abstract
The most common hemolytic anemia in the world population is sickle cell anemia, with an incidence of 1/600 newborns in the United States and Spain some regions 1/5000 incidence of infants; in Colombia there are no records regarding the incidence and prevalence.
ACF transmission is autosomal dominant. Homozygotes (SS) do not synthesize Hb A and possess erythrocytes with 90 % Hb S. The carrier or heterozygous (AS) is greater Hb RBCs with 50 % A and Hb S of 20 - 40 % and are usually asymptomatic.
Hb S is due to a mutation in the gene for beta globin chain, leading to polymerization of Hb in low oxygenation, resulting in a change in morphology sickle erythrocyte acquiring form.
The symptoms are secondary to chronic hemolytic anemia, vaso-occlusion in the different organs and functional asplenia which predisposes to infection. Other associated manifestations are splenic sequestration, erythroid aplasia complications and organ - specific, which decrease the quality of life and predispose to increased mortality.
Its management must be performed in reference centers where there is a comprehensive management including human and physical resources, as improper handling and its complications decreased survival which is not more than 45 years according to reports.
Keywords: hemolytic anemia, sickle cell syndrome, death in childhood.
INTRODUCCIÓN
La anemia hemolítica más frecuente en la población mundial es la anemia de células falciformes (ACF) (1-3).
Las anemias hemolíticas se caracterizan por una disminución de la vida media del glóbulo rojo inferior a 120 días, con un aumento de la eritropoyesis a nivel medular, que se expresa por incremento de reticulocitos en sangre periférica (2,4,5). Estos son un índice de respuesta medular con valores normales de 40 000 a 75 000, y cuando son superiores a 100 000 se asocian a hemolisis o sangrado (6).
La hemolisis puede ser extravascular, como ocurre de manera fisiológica cuando el glóbulo rojo cumple su vida media y es atrapado por los macrófagos esplénicos, así como en algunas anemias hemolíticas asociadas a defectos de membrana y las hemoglobinopatías; o intravascular, cuando los eritrocitos son destruidos en la circulación, como en las transfusiones incompatibles, enzimopatías o el síndrome hemolítico urémico (5, 7-9).
Existen diferentes variantes de hemoglobina (Hb) tantas como letras del abecedario y por nombres, según el sitio de hallazgo (10). La hemoglobina está formada por 4 cadenas de globina, cada una con un grupo hem y un átomo de hierro central (11-13). Hay 6 tipos de cadenas de globina humana: alfa, beta, delta, épsilon, zeta y gamma, y de las combinaciones dos a dos se forman los diferentes tipos de Hb; las combinaciones 2:2 de cadenas de globina alfa y beta constituyen la Hb A, que corresponde al 97 % en el niño mayor de 1 año, la Hb A2, constituida por la proporción 2:2 de cadenas alfa y delta, con un 1 a 3.5 %, y la Hb fetal (F), formada por 2 alfa y 2 gamma, que corresponde al 1 % o menos del total; esta última es mayor en el menor de un año (4,11,14).
Las anemias hemolíticas pueden ser corpusculares o intrínsecas, usualmente congénitas, como las anormalidades en la membrana, hemoglobinopatías y defectos enzimáticos; y extracorpusculares o extrínsecas, generalmente adquiridas por mecanismos inmunes o no inmunes (4,5,9).
En relación con la clínica de las anemias hemolíticas, es común la palidez por la anemia, la ictericia por aumento de la bilirrubina indirecta secundaria a la hemolisis temprana y la esplenomegalia (4,11,14).
La ictericia no es un hallazgo constante en la hemolisis, así como la esplenomegalia, la cual estará presente en relación con el tipo de hemolisis (11).
Las hemoglobinopatías más frecuentes son las estructurales, en las que la alteración es cualitativa por un cambio de 1 o más aminoácidos de la cadena de globina, como en la ACF; seguidas por las talasemias, caracterizadas por la disminución o ausencia total de la síntesis de una o varias cadenas de globina, que son defectos cuantitativos (11,15). Estas dos constituyen las alteraciones monogénicas más frecuentes en el mundo (4). La ACF es la más frecuente, con una incidencia de 1/600 recien nacidos en Estados Unidos y en algunas regiones de España con incidencia de 1/5000 neonatos; en Colombia no hay registros respecto a la incidencia y prevalencia (14).
La transmisión de la ACF es autosómica dominante (4,14). Los homocigotos (SS) no sintetizan Hb A y poseen eritrocitos con un 90 % de Hb S. El portador o heterocigoto (AS) tiene hematíes con Hb A mayor que 50 % y Hb S de 20 - 40% y son usualmente asintomáticos (5,16).
La Hb S se debe a una mutación en el gen de la cadena beta de globina, en el cromosoma 11, en su sexto codón, donde se sustituye adenina por timina, lo cual genera el cambio de ácido glutámico por valina; esto conlleva a la polimerización de la Hb en condiciones de baja oxigenación; debido a lo cual se origina un cambio en la morfología del eritrocito, que adquiere la forma falciforme (4,5,9,11,14,17,18).
Se sabe que la mutación surgió de manera independiente en 5 ocasiones diferentes, hace unos 4000 años, y en relación con la malaria. Estos polimorfismos modulan la expresión de la enfermedad. En África son cuatro: Benín 50-70 % de la población con Hb S africana; Bantú, república centroafricana, del 15 al 30 %, Senegal, 5 a 15 %, y Camerún. El Bantú es el que representa mayor gravedad.
Surgió una quinta mutación, la indo-arábica o haplotipo asiático, caracterizado por altos niveles de Hb F y curso leve.
La Hb S confiere protección al paludismo por Plasmodium Falciparum y aumenta la supervivencia en portadores (4, 11, 19, 20).
FISIOPATOLOGÍA
El evento primordial de la clínica y complicaciones en el paciente con ACF son producidas por la vaso - oclusión y la isquemia tisular secundaria (4,11,14).
La Hb S a bajas concentraciones de oxígeno es menos soluble que la Hb A, lo cual genera polímeros y rigidez del eritrocito, que son menos deformables y causan su destrucción precoz. Los cambios constantes de oxigenación-desoxigenación producen lesión en la membrana del hematíe, alteración de la bomba de iones y deshidratación celular; lo anterior condiciona a destrucción intravascular temprana. Esta hemolisis genera reducción del óxido nítrico y conlleva a vaso-oclusión y activación plaquetaria. La exposición de la fosfatidilserina secundario al daño de membrana activa la cascada de la coagulación y se produce un incremento en la viscosidad sanguínea local (21).
Otros factores que influyen en la vaso-oclusión e isquemia son la activación del endotelio, el incremento de las propiedades adhesivas de células rojas y leucocitos reducen el flujo sanguíneo. Además, la leucocitosis y trombocitosis habitual aumentan la viscosidad sanguínea (5, 11, 14, 17, 22-26).
La Hb S puede polimerizar con otros tipos de Hb; la Hb F es con la que polimeriza menos; esto es un mecanismo protector en los pacientes con ACF que condiciona una mayor o menor severidad (4,14).
DIAGNÓSTICO
El hemograma en el SS muestra anemia normocítica normocrómica, usualmente leucocitosis y trombocitosis, asociados a reticulocitosis. Si es S-beta talasemia, habrá microcitosis. La sedimentación globular estará disminuida por la rigidez de los glóbulos rojos. Es usual en sangre periférica la policromatofilia, normoblastos, células falciformes y dianocitos.
La siclemia es una prueba de tamizaje con metabisulfito de sodio que indica la presencia de células falciformes pero no hace diagnóstico y es poco útil en recien nacidos y lactantes por el alto contenido de Hb F.
La electroforesis de hemoglobina en el recién nacido es ASF, en el mayor de 1 año AS en los heterocigotos y en el homocigoto será SS o SF. Ver tabla 1 para los diferentes síndromes falciformes (4, 5, 22, 27).
MANIFESTACIONES CLÍNICAS
La sintomatología es secundaria a la anemia hemolítica crónica, la vaso-oclusión en los diferentes órganos y la asplenia funcional, la cual predispone a la infección, y es una causa importante de muerte en la edad pediátrica. Otras manifestaciones asociadas son el secuestro esplénico, la aplasia eritroide y las complicaciones órgano - especificas, que disminuyen la calidad de vida y predisponen a mayor mortalidad.
Usualmente el paciente sin crisis presenta palidez, ictericia, esplenomegalia, peso bajo por la hipoxia y niveles de Hb entre 6 y 8 gr/dl. La Hb S tiene poca afinidad por el oxígeno y no siempre hay sintomatología de anemia con niveles bajos de Hb; sin embargo, se debe evitar la sobrecarga de volumen por el riesgo de Cor anémico (4,5,25,28).
APLASIA PURA TRANSITORIA ERITROIDE
Es el freno en la producción de glóbulos rojos a nivel medular, asociado a infección generalmente por parvovirus B19 y se presenta a cualquier edad. Se caracteriza por disminución brusca de la Hb, usualmente inferior a 5 gr, y puede descender hasta 1 gramo, con reticulocitopenia. El recuento de leucocitos y plaquetas son normales.
Esta puede persistir por 10 a 14 días; su manejo es la observación, el ácido fólico para prevenir la megaloblastosis y la transfusión, si se requiere (4, 14, 29).
SECUESTRO ESPLÉNICO
Se presenta entre los 6 y 24 meses de edad, usualmente puede ser fatal. Se caracteriza por esplenomegalia progresiva y masiva con atrapamiento de grandes cantidades de sangre en el bazo, lu cual produce dolor abdominal, náuseas y vómitos. El nivel de Hb puede caer bruscamente, seguido por shock hipovolémico y muerte.
En el hemograma hay anemia, y puede haber trombocitopenia asociada. Se sospecha cuando aumenta la necesidad de transfusión en forma repetida cada 30 a 45 días, debido a que estos pacientes requieren en promedio de 2 a 3 transfusiones al año.
La esplenectomía es el tratamiento de elección si el episodio es mayor, asociado a falla multisistémica o más de dos episodios menores (5, 11, 14, 29, 30).
CRISIS HIPERHEMOLÍTICAS
A pesar de ser una anemia hemolítica, estas crisis han sido descritas asociándolas a infecciones y ciertos medicamentos, o durante una crisis vaso-oclusiva severa, los cuales aumentan la destrucción del glóbulo rojo, con una marcada disminución de la hemoglobina, reticulocitosis y exacerbación de la ictericia (4, 5, 14, 28).
CRISIS VASO - OCLUSIVAS
Son las que generan las principales manifestaciones y complicaciones de esta patología. Entre las crisis óseas encontramos la dactilitis, que es la primera manifestación en el lactante y se caracteriza por edema del dorso de manos y pies, dolor, fiebre, derrame articular, leucocitosis con neutrofília, disminución del movimiento secundaria al compromiso de las falanges, llanto e irritabilidad (4, 5). Es más común en las estaciones frías y asociada a infecciones. Con el tiempo el tejido medular rojo es reemplazado por tejido fibroso, el cual presenta menos demanda de oxígeno y puede perdurar en situaciones de anaerobiosis (16). Por la injuria continua aparecen los dedos marfanoides o bradidactilia, además de las epífisis en cono y metáfisis con cavidades (31).
Las crisis de dolor óseo, características de esta patología, son las más frecuentes, representadas por la isquemia de la médula ósea. Afecta usualmente a huesos largos y también planos. Si hay fiebre, se deben cultivos para descartar infección por Staphylococcus Aureus y Salmonella (5, 14, 32, 33).
Los dolores óseos son 50 veces o más frecuentes que la osteomielitis (16). La intensidad, duración y características del dolor son variables, comprometen múltiples sitios a la vez. En caso de que se sospeche de infección, debe completarse el estudio con imágenes diagnósticas y pruebas bacteriológicas que pueden incluir la punción (31, 34).
Cuando los dolores óseos mejoran y de manera residual persiste dolor óseo único con o sin signos inflamatorios, podemos estar ante la presencia de una infección o un infarto óseo (14, 35).
El tratamiento fundamental es la hidratación, evitando la sobrecarga de volumen, cuya tasa hídrica dependerá del nivel de hemoglobina. Los dolores leves pueden ser tratados ambulatoriamente con líquidos orales, acetaminofén e ibuprofeno (14,16). Si el dolor es moderado a severo, es prioritaria la hospitalización para evitar mayor isquemia tisular a órganos blancos, y su manejo incluye líquidos parenterales a 1800 cc/m2sc/día o menos inicialmente, dependiendo del nivel de hemoglobina, analgésicos por horario, como el tramadol 1 a 2 mg/kg/dosis intravenoso cada 6-8 horas, alternando con acetaminofén 15 mg/kg/dosis cada 6 - 8 horas o ibuprofeno 5 - 10 mg/kg/dosis cada 6 a 8 horas. No se debe suministrar analgésicos con dosis - respuesta si no se logra el control del dolor (5, 14, 33, 36-38). Si es refractaria a este manejo, se debe considerar la transfusión de hematíes.
En urgencias se debe tener presente que hidratar, suministrar analgesia, controlar dolor y dar manejo ambulatorio en casos de dolores severos, en vez del manejo intrahospitalario descrito, aumenta la tasa de reingresos, y es más complejo su control y requiere mayor estancia, medicamentos, transfusiones y riesgo de secuelas con ausencia escolar y mala calidad de vida (5,14,37,39,40).
La necrosis avascular es más frecuente en la cabeza femoral y en la segunda década de la vida, aunque puede encontrarse en muñeca, y es menos común en los huesos tubulares largos. Hay reportes de osteonecrosis en el talus y calcáneo (4). Es común en los SS, hemoglobinopatías S-alfa talasemia y en la SC. La cabeza femoral, al ser parte de una articulación de carga, recibe el peso corporal, debido a lo cual se produce una obstrucción de los sinusoides medulares, necrosis y colapso. El 50 % pueden ser asintomáticos, pero la progresión genera dolor y alteración de la marcha, con limitación de la rotación interna (4, , 14).
El diagnóstico temprano se hace con la resonancia magnética, detectando los cambios óseos incipientes en pacientes asintomáticos o con poca clínica. El manejo general inicialmente incluye ortopedia y reposo de la articulación para limitar el movimiento, antiinflamatorios no esteroideos y por último descompresión y prótesis (4, 5, 14).
SÍNDROME DE TÓRAX AGUDO (STA)
Tiene su mayor incidencia en la adolescencia y con la infección son causas frecuentes de muerte y hospitalización. Debe sospecharse ante dolor torácico, asociado a tos, disnea, fiebre, y es indistinguible de una neumonía. Su etiología más frecuente en niños son las infecciones por Mycoplasma Pneumoniae, Chlamydia Pneumoniae, seguida por Neumococo, Parvovirus B 19 y otros virus (5,14). Otras causas son la embolización grasa secundaria a necrosis de la médula ósea posterior a una crisis vaso-oclusiva, por oclusión vascular pulmonar (trombosis in situ) y por hipoventilación con hipoxemia y vaso-oclusión subsiguiente como en el infarto costal o en la anestesia general (5).
Su tratamiento incluye hidratación, analgesia, antibioticoterapia de amplio espectro (cefalosporinas y macrólidos), oxigenoterapia, que puede requerir ventilación mecánica, broncodilatadores, dexametasona y transfusión o exanguinotransfusión (5,14).
DOLOR ABDOMINAL
El dolor abdominal es una manifestación común que puede semejar un abdomen agudo quirúrgico. Puede producirse por obstrucción de vasos mesentéricos o por infarto hepático, esplénico o de nódulos linfáticos (4,14).Otras causas son la necrosis papilar renal, infarto vertebral y costal. Una causa importante es la colelitiasis, que se puede presentar en edades tempranas. El dolor abdominal con fiebre, ictericia obstructiva y aumento de enzimas hepáticas lleva diagnosticar de colestasis intrahepática (4, 5).
PRIAPISMO
Del griego Priapus, que era el dios de la fertilidad y la suerte. Es una erección mantenida y dolorosa. Se produce por obstrucción del flujo de salida venosa e ingurgitación secundaria de los cuerpos cavernosos. La consulta a urgencias se estima en un 30 %, aunque podría ser superior, sin embargo, en muchos casos no consultan por prejuicio social. La erección fisiológica es usual en la madrugada por la deshidratación y la acidosis, con aumento de la polimerización y rigidez del eritrocito. Como factores predisponentes se encuentran la masturbación, infección y trauma local (4, 5, 41, 42).
Pueden ser episodios cortos, múltiples e intermitentes durante 3 horas, generalmente nocturnos, que aparecen como premonitorios de episodios largos, Su tratamiento es la hidratación oral, analgésicos, micciones frecuentes, ejercicios, baños con agua caliente y el apoyo de urología.
Los episodios prolongados mayores de 3 horas pueden ser concomitantes a manifestaciones neurológicas y cuando persisten más de 4 horas pueden generar injuria irreversible y disfunción sexual (4,5,14). Su tratamiento es intrahospitalario con líquidos endovenosos, analgesia, aspiración del cuerpo cavernoso, seguido de irrigación con adrenalina (1:1.000.000), o incluso una infusión con fenilefrina y un agente alfa adrenérgico. Considerar la exanguinotransfusión si persiste, la cual se ha asociado a eventos neurológicos. Si continúa se debe realizar derivación cavernosa - esponjosa o cavernosa - safena. Se han descrito fármacos que eviten la recurrencia temprana, como la pseudoefedrina, hidroxiurea, inyecciones de leuprolide, además de un protocolo de transfusiones por 6 a 12 meses (5, 14).
ENFERMEDAD CEREBRO-VASCULAR (ECV)
Aunque frecuente, no tiene una alta mortalidad, pero sus secuelas generan deterioro de la calidad de vida. Se produce por el daño de la célula falciforme a la íntima vascular, lo cual genera proliferación de fibroblastos y musculo liso, lo cual se produce estrechez del lumen de las arterias cerebrales, y esto, un aumento de la velocidad de flujo, dejando zonas isquémicas con posterior revascularización. Esta angiogénesis posterior se denomina enfermedad de moyamoya, la cual se confirma con la angioresonancia de cerebro (5,14,43-45).
La forma isquémica es la más frecuente, con una prevalencia del 11 % antes de los 20 años, y mayor en la primera década, con 1.02 por cada 100 niños de 2 a 5 años y 0.79 por cada 100 niños de los 6 a 9 años (5).
Los vasos más comprometidos son la arteria carótida interna y la cerebral media izquierdas. Pueden haber episodios asintomáticos o infartos silenciosos en 20 a 30 %, que se diagnostican por resonancia magnética de cerebro, porque son microvasculares, y el doppler transcraneal (DTC) no es útil, debido a que este último detecta en mayor proporción las macrovasculopatías (5, 14, 46, 47).
Los síntomas incluyen cefalea, convulsiones, hemiparesia, alteración en la marcha, habla, del estado mental y cognitivo. Puede haber recuperación motora, pero el déficit neurocognitivo habitualmente persiste (5, 48).
Su tratamiento es la exanguinotransfusión automatizada (eritrocitoaféresis) o manual (flebotomias) para reducir la Hb S un 30 % o menos y elevar la Hb a 10 gr/dl. Si no es posible lo anterior, realizar la transfusión de eritrocitos para alcanzar el mismo nivel de Hb.
Por tener recurrencia elevada, el paciente debe ingresar a transfusiones crónicas de manera indefinida y debe acompañarse de rehabilitación (5,14,16).
Otras alternativas son la estimulación de la Hb fetal, trasplante de precursores hematopoyéticos y revascularización.
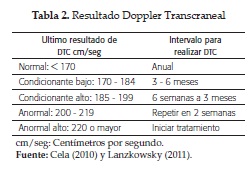
Para evaluar el riesgo y prevenir la enfermedad cerebrovascular isquémica se utiliza el Doppler Transcraneal, el cual mide la velocidad del flujo en las arterias cerebrales grandes y debe realizarse periódicamente entre los 2 y 16 años. Su valor normal es de 170 cm/seg o menor. Velocidades inferiores a 70 cm/seg indican estenosis severa y riesgo aumentado de ECV (tabla 2).
INFECCIÓN
Es la causa más frecuente de muerte en los primeros 5 años. Es secundaria al hipoesplenismo funcional causado por el siclaje intraesplénico que conduce a fibrosis progresiva y autoesplenectomia, la cual favorece las infecciones por gérmenes encapsulados y riesgo de sepsis.
Las infecciones por Neumococo son 300 a 600 veces más frecuentes (5,14).
En menores de 5 años con fiebre debe realizarse hemograma, hemocultivo, uroanalisis, urocultivo, radiografía de tórax e iniciar cefalosporina de tercera generación; pero si hay compromiso pulmonar, adicionar claritomicina (5, 14, 32, 49).
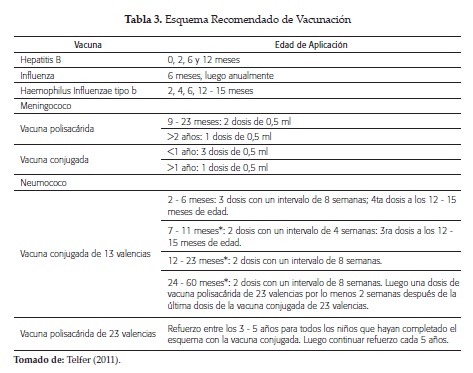
Todos los pacientes deben recibir profilaxis con penicilina oral desde los 3 meses, a dosis de 250 mg/día, hasta los 3 años, y después 500 mg/día hasta los 5 años. Por lo anterior requiere el concurso de infectologia e inmunización adecuada (4, 5) (tabla 3).
Las complicaciones crónicas deterioran la calidad de vida e incluyen compromiso de órganos como corazón, cerebro, pulmones, riñones, hígado, ojos, piel, entre otros. Es una enfermedad silente en muchos de ellos, pero con complicaciones graves e irreversibles al diagnóstico.
En relación con el desarrollo pondoestatural y gonadal, se comprometen desde el segundo año de vida, aplanando la curva. Al final de la adolescencia puede recuperarse la altura pero no el peso de los controles sanos.
La maduración sexual máxima se alcanza a los 17 años en promedio.
El Sulfato de Zinc, a dosis de 220 mg, 3 veces al día desde los 10 años, ha mostrado mejoría significativa en el crecimiento y desarrollo gonadal (5,14).
TRATAMIENTO
Para estapatología se recomienda que se realize en centros de referencia para esta patología donde haya un manejo integral, incluyendo el recurso humano y físico, ya que el manejo inadecuado y sus complicaciones disminuyen la sobrevida, la cual no es superior a los 45 años según reportes.
El diagnóstico temprano permite un mejor manejo en la edad pediátrica y disminuye o evita complicaciones tardías (5).
Todo procedimiento quirúrgico es de riesgo mayor y los pacientes deben ser hospitalizado previamente para evaluar necesidad de transfusión, Doppler Transcraneal reciente, hidratación, antibióticos y valoración por anestesia.
Factores como el frío, deshidratación, estrés, hipoxia, infección, trauma y medicamentos están asociados a polimerización y vaso-oclusión (5).
Las cirugías más frecuentes en estos pacientes son la colecistectomía por colelitiasis, esplenectomía en secuestros esplénicos graves o repetidos, adenoidectomía y amigdalectomía en hipertrofias con obstrucción de vía aérea y colocación de catéteres centrales permanentes (4, 5).
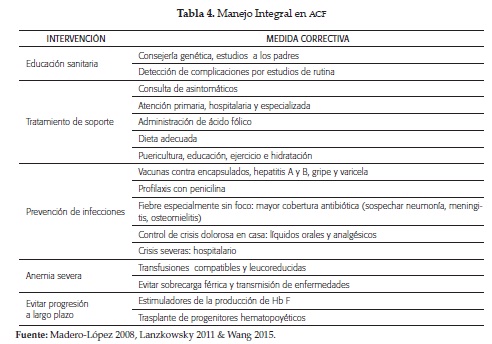
El tratamiento es integral (tabla 4), y para el seguimiento del paciente se recomienda estudios en forma periódica (tabla 5). Durante la revisión se explicó el manejo de los eventos más importantes. Otras modalidades terapéuticas son las transfusiones, hidroxiurea y trasplante (4, 5, 14, 50).
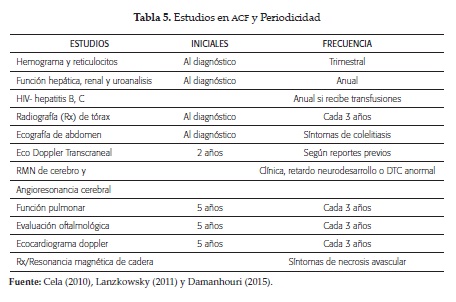
TRANSFUSIONES
Usualmente estos pacientes requieren transfusión de hematíes con niveles de Hb de 5 gr/dl o inferiores si no están asociados a un evento agudo o una complicación crónica (tabla 6) (5,14).
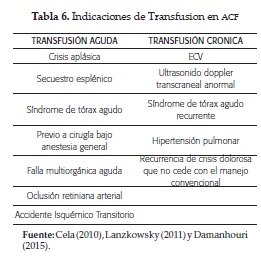
La aloinmunización en ACF es aproximadamente del 17 % y la mayor frecuencia es a Kell y Rh (C y E). Idealmente debe realizarse fenotipo de células rojas al momento del diagnóstico y los concentrados leucoreducidos (51, 52).
Existen tres tipos de modalidades transfusionales: las simples, utilizadas en secuestro esplénico, aplasia eritroide, previo a cirugía e insuficiencia cardiaca. Exanguinotransfusión en crisis graves como ECV, STA con mala evolución, priapismo y crisis de dolor refractarias, la cual se puede realizar en forma automatizada o manual; y esta última por vía central o periférica.
Todas las extracciones y transfusiones pueden ser realizadas por la misma vía. Con Hb entre 8 y 9 gr/dl se remueven 5 cc/kg de sangre, luego se infunden 5 cc/kg de solución salina 0,9% (SSN), y posteriormente se transfunde hematíes a 15 cc/kg.
Cuando la Hb es mayor que 9 gr/dl se practica flebotomía de 5 cc/kg de sangre con infusión posterior de 10 cc/kg de SSN, una segunda flebotomía de 5 cc/kg y transfusión de 15 cc/kg de hematíes. La última modalidad es la profiláctica, periódica, generalmente mensual, ante riesgo de ECV, STA a repetición y ulceras crónicas (5, 16).
SOBRECARGA DE HIERRO
Consecuencia de regímenes transfusionales crónicos que conllevan a la acumulación temprana de hierro en la infancia, lo cual produce daño cardiaco, hepático y de glándulas endocrinas.
El monitoreo de la sobrecarga se puede realizar por la medición de los niveles de ferritina sérica y concentración de hierro en hígado o corazón por resonancia magnética.
Otra manera menos utilizada es la biopsia hepática, indicada cuando se requiere diagnostico histológico.
Recientemente se está introduciendo la medición de fracciones de hierro no unido a transferrina, que representa el hierro plasmático lábil responsable de la acción catalítica oxidativa por la formación de radicales libres a nivel celular hepático, cardiaco y endocrino (4,14).
Las indicaciones de quelación del hierro son una carga transfusional acumulada de 120 cc/kg o mayor, niveles de ferritina de 1000 ng/ml o mayor y concentración hepática de hierro mayor que 5-7 mg/gr de peso seco (14).
El único quelante oral en nuestro medio es el deferasirox, y los estudios han mostrado eficacia similar a la deferoxamina utilizada por vía subcutánea o endovenosa. Aún faltan estudios que demuestren la eficacia para remover el hierro cardiaco. La dosis es 20 - 40 mg/kg/día, su vida media de 12 - 16 horas con eliminación intestinal. Su presentación es en tabletas de 250 y 500 mg. Tiene mínimos efectos adversos, sobre todo gastrointestinal; además se ha descrito elevación de transaminasas, rash y aumento de creatinina (53).
HIDROXIUREA
Los pacientes con niveles de Hb F presentes o aumentados se asocian a menos vaso-oclusión. La hidroxiurea es un estimulador de la producción de Hb F, pero requiere varios meses para obtener el efecto deseado. Se caracteriza por su rápida absorción, alta biodisponibilidad y adecuada tolerancia. Otros efectos mediatos son la hidratación celular, aumento de óxido nítrico, disminución de leucocitos, plaquetas y adhesividad del hematíe (4, 5, 14, 16, 46, 54, 55).
Las indicaciones son: tres o más eventos vaso-oclusivos severos en 1 año y que usualmente ameriten hospitalización, prevención de un STA recurrente, priapismo persistente sin respuesta a manejo convencional, prevención de ECV recurrente y con Doppler Transcraneal anormal asociados o no a transfusiones. Hay estudios recientes de su utilización en Doppler anormal para evitar progresión a ECV (5,44,55).
La dosis inicial es de 15 - 20 mg/kg/día por vía oral y se incrementa 5 mg/kg/día cada 8 semanas hasta una dosis máxima de 35 mg/kg/día. Su presentación es en tabletas de 500 mg. La toxicidad es infrecuente y se sospecha cuando los neutrófilos son < 1000/mm3, plaquetas < 80000/mm3, caída de hemoglobina 2 gr/dl y reticulocitos < 80000 /mm3. Se han reportado efectos adversos en el crecimiento, desarrollo y teratógenos, además de caída del cabello, cambios pigmentarios en piel, alteraciones gastrointestinales y reducción de conteo espermático y motilidad (5).
Realizar hemograma cada 4 semanas hasta estabilizar y luego cada 8 -12 semanas.
Otros moduladores son los butiratos y la decitabina (14, 56).
Conflicto de intereses: ninguno.
Financiación: Universidad Simón Bolívar
REFERENCIAS
1. Loureiro MM, Rozenfeld S. Epidemiologia de internações por doença falciforme no Brasil. Rev Saude Publica 2005;39(6):943-9. Disponible en: http://dx.doi.org/10.1590/S0034-89102005000600012. [ Links ]
2. García-Rodríguez MJ, Álvarez ER, Morado Arias M, Hernández-Navarro F. Protocolo diagnóstico de las anemias hemolíticas. Medicine 2008;10(20):1371-4. [ Links ]
3. Piel FB, Hay SI, Gupta S, Weatherall DJ, Williams TN. Global Burden of Sickle Cell Anaemia in Children under Five, 2010-2050: Modelling Based on Demographics, Excess Mortality, and Interventions. Plos Medicine 2013;10(7):e1001484 http://dx.doi.org/10.1371/journal.pmed.1001484. [ Links ]
4. Madero-López L, Muñoz Villa A. Hematología y oncología pediátricas. 2a ed. Madrid: Ergon; 2005. [ Links ]
5. Lanzkowsky P. Classification and Diagnosis of Anemia in Children. En Manual of Pediatric Hematology and Oncology. Elsevier 2011. p. 1-13. [ Links ]
6. Badami KG. The reticulocyte count. Natl Med J India 1990;3(2):69-72. [ Links ]
7. Clinton-Hidalgo JA. Síndrome de anemia hemolítica (Revisión bibliográfica). Rev Medica Costa Rica y Centroam 2008;LXV(583):85-90. Disponible en: http://www.binasss.sa.cr/revistas/rmcc/583/art2.pdf. [ Links ]
8. Mejía-Arreguí MH. Anemias hemolíticas autoinmunes. Rev Med Inst MexSeguro Soc 2005;43(supl1):25-8. Disponible en: http://www.medigraphic.com/pdfs/imss/im-2005/ims051g.pdf. [ Links ]
9. González-García H. Anemias hemolíticas en la infancia. Pediatr Integr 2012;16(5):378-86. [ Links ]
10. Fundatal FI de T. Guidelines for the Clinical Management of Thalassaemia. 2a ed. Team Up Creations Ltd, editor. Nicosia, Chipre: Federación Internacional de Talasemia; 2008. [ Links ]
11. Orkin SH, Nathan DG, Ginsburg D, Look AT, Fisher DE, Samuel LI. Nathan and Oski's Hematology of Infancy and Childhood: Expert Consult. 2012. [ Links ]
12. Guyton, CG and Hall, JE. Tratado de Fisiología Médica. 11ª ed. Elsevier, 2006. [ Links ]
13. Sarnaik SA. Thalassemia and related hemoglobinopathies. Indian J Pediatr 2005; 72(4):319-24. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15876761. [ Links ]
14. Madero-López L, Muñoz Villa A. Hematología y oncología pediátrica. 3a ed. Madrid: Ergon; 2008. [ Links ]
15. Jacobsen ED, White BM, White BT, Batt K, Reske T. Hematology Board Review Manual Hemoglobinopathies. Hematology 2010;5(3):1-12. [ Links ]
16. Cela E, Cervera Á, Díaz de Heredia C, Rives S, Salinas JA, Sevilla J et al. Guía de práctica clínica sobre enfermedad de células falciformes pediátrica. Sehop, editor. SEHO, Sociedad Española de Hematología y Oncología Pediátricas; 2010. [ Links ]
17. Odièvre M, Verger E, Silva-Pinto AC, Elion J. Pathophysiological insights in sickle cell disease. Indian J Med Res 2011;134:532-7. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/22089617 [ Links ]
18. McCavit TL. Sickle Cell Disease. Pediatr Rev 2012;33(5):195-206. Disponible en: http://pedsinreview.aappublications.org/cgi/doi/10.1542/pir.33-5-195. [ Links ]
19. Rodríguez-Romero WE, Sáenz-Renauld GF, Chaves-Villalobos MA. Haplotipos de la hemoglobina S: importancia epidemiológica, antropológica y clínica. Rev Panam Salud Pública 1998;3(1):1-8. [ Links ]
20. Makani J, Ofori-Acquah SF, Nnodu O, Wonkam A, Ohene-Frempong K. Sickle Cell Disease: New Opportunities and Challenges in Africa. Sci World J 2013;2013:1-16. Disponible en: http://dx.doi.org/10.1155/2013/193252. [ Links ]
21. Pakbaz Z, Wun T. Role of the Hemostatic System on Sickle Cell Disease Pathophysiology and Potential Therapeutics. Hematol Oncol Clin North Am. 2014;28(2):355-74. Disponible en: http://dx.doi.org/10.1016/j.hoc.2013.11.011. [ Links ]
22. Jain S, Kapetanaki MG, Raghavachari N, Woodhouse K, Yu G, Barge S et al. Expression of Regulatory Platelet MicroRNAs in Patients with Sickle Cell Disease. Chi J-TA, editor. PLoS One 2013;8(4):e60932. Disponible en: http://dx.plos.org/10.1371/journal.pone.0060932. [ Links ]
23. Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet 2010; 376(9757):2018-31. Disponible en: http://dx.doi.org/10.1016/S0140-6736(10)61029-X. [ Links ]
24. Hebbel RP, Osarogiagbon R, Kaul D. The Endothelial Biology of Sickle Cell Disease: Inflammation and a Chronic Vasculopathy. Microcirculation 2004;11(2):129-51. Disponible en: http://doi.wiley.com/10.1080/10739680490278402. [ Links ]
25. Abboud MR, Musallam KM. Sickle cell disease at the dawn of the molecular era. Hemoglobin 2009; 33 Supl 1:S93-106. Doi: 10.3109 / 03630260903347617. [ Links ]
26. Wood KC, Hsu LL, Gladwin MT. Sickle cell disease vasculopathy: A state of nitric oxide resistance. Free Radic Biol Med 2008; 44(8):1506-28. Doi: 10.1016 / J.Freeradbiomed.2008.01.008 [ Links ]
27. Damanhouri GA, Jarullah J, Mushtaq G, Kamal MA. Clinical biomarkers in sickle cell disease. Saudi J Biol Sci King Saud University 2015;22(1):24-31. Disponible en: http://dx.Doi.org/10.1016/j.sjbs.2014.09.005. [ Links ]
28. Conran N, Franco-Penteado CF, Costa FF. Newer Aspects of the Pathophysiology of Sickle Cell Disease Vaso-Occlusion. Hemoglobin 2009; 33(1):1-16. Doi:10.1080 / 03630260802625709 [ Links ]
29. Ballas SK. Defining the Phenotypes of Sickle Cell Disease. Hemoglobin 2011; 35(5-6):511-9. Doi:10.3109/03630269.2011.610477 [ Links ]
30. Vick LR, Gosche JR, Islam S. Partial splenectomy prevents splenic sequestration crises in sickle cell disease. J Pediatr Surg 2009; 44(11):2088-91. Disponible en: http://dx.Doi.org/10.1016/j.jpedsurg.2009.06.007. [ Links ]
31. Kupersmith LM, Norton KI, Hausman MR. Sickle cell anemia and the hand. J Am Soc Surg Hand 2003; 3(3):145-51. Disponible en: http://dx.Doi.org/10.1016/S1531-0914(03)00068-8. [ Links ]
32. Booth C, Inusa B, Obaro SK. Infection in sickle cell disease: A review. Int J Infect Dis 2010; 14(1):e2-12. Doi: http://dx.doi.org/10.1016/j.ijid.2009.03.010. [ Links ]
33. Niscola P, Sorrentino F, Scaramucci L, De Fabritiis P, Cianciulli P. Pain Syndromes in Sickle Cell Disease: An Update. Pain Med 2009;10(3):470-80. Disponible en: http://dx.doi.org/10.1111/j.1526-4637.2009.00601. [ Links ]
34. Ohara DG, Ruas G, Castro SS, Martins PRJ, Walsh IAP. Musculoskeletal pain, profile and quality of life of individuals with sickle cell disease. Rev Bras Fisioter 2012;16(5):431-8. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/22886311. [ Links ]
35. Quirolo K. How do I transfuse patients with sickle cell disease? Transfusion 2010;50(9):1881-6. Doi: 10.1111/j.1537-2995.2010.02774.x [ Links ]
36. Myers M, Eckes EJ. A novel approach to pain management in persons with sickle cell disease. Medsurg Nurs 2012;21(5):293-8. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/23243787. [ Links ]
37. Erhan E, Inal Mt, Aydinok Y, Balkan C, Yegul I. Tramadol infusion for the pain management in sickle cell disease: a case report. Pediatr Anesth 2007;17(1):84-6. Disponible en: http://doi.wiley.com/10.1111/j.1460-9592.2006.02030.x. [ Links ]
38. Ballas SK. Update on Pain Management in Sickle Cell Disease. Hemoglobin 2011;35(5-6):520-9. Doi:10.3109 / 03630269.2011.610478 [ Links ]
39. Zempsky WT, Loiselle KA, McKay K, Lee BH, Hagstrom JN, Schechter NL. Do Children with Sickle Cell Disease Receive Disparate Care for Pain in the Emergency Department? J Emerg Med 2010;39(5):691-5. Disponible en: http://dx.doi.org/10.1016/j.jemermed.2009.06.003. [ Links ]
40. Kavanagh PL, Sprinz PG, Vinci SR, Bauchner H, Wang CJ. Management of Children With Sickle Cell Disease: A Comprehensive Review of the Literature. Pediatrics 2011;128(6):e1552-74. Disponible en: http://pediatrics.aappublications.org/cgi/doi/10.1542/peds.2010-3686. [ Links ]
41. Roizenblatt M, Figueiredo MS, Cançado RD, Pollack-Filho F, de Almeida Santos Arruda MM, Vicari P et al. Priapism is Associated with Sleep Hypoxemia in Sickle Cell Disease. J Urol 2012;188(4):1245-51. Disponible en: http://dx.doi.org/10.1016/j.juro.2012.06.015. [ Links ]
42. Chrouser KL, Ajiboye OB, Oyetunji TA, Chang DC. Priapism in the United States: the changing role of sickle cell disease. Am J Surg 2011;201(4):468-74. Disponible en: http://dx.doi.org/10.1016/j.amjsurg.2010.03.017. [ Links ]
43. Vermylen C. Sickle cell anaemia: Current therapies. Transfus Apher Sci 2013;49(2):151-4. Disponible en: http://dx.doi.org/10.1016/j.transci.2013.07.018. [ Links ]
44. Farina FM, Rampazzo P, Sainati L, Manara R, Onofri A, Colombatti R, et al. Transcranial Doppler sonography in children with sickle cell disease and silent ischemic lesions. Perspect Med 2012;1(1-12):269-71. Disponible en: http://dx.doi.org/10.1016/j.permed.2012.02.046. [ Links ]
45. Fryer RH, Anderson RC, Chiriboga CA, Feldstein NA. Sickle cell anemia with moyamoya disease: outcomes after EDAS procedure. Pediatr Neurol 2003;29(2):124-30. Disponible en: http://dx.doi.org/10.1016/S0887-8994(03)00047-X. [ Links ]
46. Telfer PT. Management of sickle cell disease: out-patient and community aspects. Paediatr Child Health (Oxford) [en línea]. Elsevier Ltd; 2011 Aug;21(8):357-62. Disponible en: http://dx.doi.org/10.1016/j.paed.2011.03.005. [ Links ]
47. Wong W, Powars DR. Overt and Incomplete (Silent) Cerebral Infarction in Sickle Cell Anemia: Diagnosis and Management. Neuroimaging Clin N Am 2007; 17(2):269-80. Disponible en: http://linkinghub.elsevier.com/retrieve/pii/S105251490700024X. [ Links ]
48. Morton C, Key N. Stroke in sickle cell disease. Semin Cerebrovasc Dis Stroke 2002;2(2):143-50. Doi:10.1053/Scds.2002.32659 [ Links ]
49. Shihabuddin BS, Scarfi CA. Fever in Children With Sickle Cell Disease: Are All Fevers Equal? J Emerg Med [en línea]. 2014 Oct;47(4):395-400. Disponible en: http://dx.doi.org/10.1016/j.jemermed.2014.06.025. [ Links ]
50. Wang WC. Newborn screening for sickle cell disease: necessary but not sufficient. J Pediatr 2015;91(3):210-2. Disponible en: http://dx.doi.org/10.1016/j.jped.2015.01.002. [ Links ]
51. Tarud GD, Prieto-Rocha P, Vives-Puello R. Hemoderivados en Recién Nacidos y Niños. Barranquilla (Colombia): Ediciones UniNorte; 2008. [ Links ]
52. Campbell-Lee SA, Kittles RA. Red Blood Cell Alloimmunization in Sickle Cell Disease: Listen to Your Ancestors. Transfus Med Hemotherapy 2014;41(6):6-6. Doi: 10.1159 / 000369513. [ Links ]
53. Marsella M, Borgna-Pignatti C. Transfusional Iron Overload and Iron Chelation Therapy in Thalassemia Major and Sickle Cell Disease. Hematol Oncol Clin North Am 2014;28(4):703-27. Disponible en: http://dx.doi.org/10.1016/j.hoc.2014.04.004. [ Links ]
54. Mohammed Z, Hawsawi A, Turkistani WA. Effect of Hydroxyurea in Children With Sickle Cell Disease in Saudi Arabia. J Taibah Univ Med Sci 2008; 3(2):129-34. Disponible en: http://dx.doi.org/10.1016/S1658-3612(08)70062-3. [ Links ]
55. Odièvre M-H, Lapouméroulie C, Elion J. Hydroxyurée et drépanocytose: rôle des protéines d'adhérence. Arch Pédiatrie 2009;16(2):95-8. [ Links ]
56. Wang WC, Helms RW, Lynn HS, Redding-Lallinger R, Gee BE, Ohene-Frempong K, et al. Effect of hydroxyurea on growth in children with sickle cell anemia: Results of the HUG-KIDS study. J Pediatr 2002;140(2):225-9. [ Links ]
57. Vermylen C. Hematopoietic stem cell transplantation in sickle cell disease. Blood Rev 2003; 17(3):163-6. [ Links ]

