










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Salud Uninorte
Print version ISSN 0120-5552On-line version ISSN 2011-7531
Salud, Barranquilla vol.32 no.3 Barranquilla Sept./Dec. 2016
Síndrome de Kabuki: presentación de un Caso y Revisión de la Literatura
Kabuki Syndrome: A case report and literature review
Carlos Osorio Alarcón1, Diego Olaya Mantilla1, Carlos Silvera Redondo1, Pilar Garavito Galofre1
1 Grupo de Investigación en Genética y Medicina Molecular, Universidad del Norte. Barranquilla. (Colombia).
Correspondencia: Pilar Garavito Galofre. Universidad del Norte. Barranquilla (Colombia). A.A. 1609. mpgaravi@uninorte.edu.co.
Fecha de recepción: 22 de agosto de 2016
Fecha de aceptación: 15 de noviembre de 2016
Resumen
El síndrome de Kabuki (SK) es una patología muy rara, descrita por primera vez en 1981 por Niikawa y Kuroki en Japón. Se han publicado cerca de 400 casos a nivel mundial. En Colombia se conocen cinco casos diagnosticados y publicados; el caso objeto de este estudio sería el sexto en nuestro país. Presentamos la descripción del caso de una paciente de 2 años y 6 meses con rasgos dismórficos compatibles con síndrome de Kabuki. Examen físico: fisuras palpebrales elongadas, eversión del tercio lateral párpado inferior, cejas arqueadas con tercio lateral más despoblado, puente nasal deprimido, boca en carpa, paladar hendido, pabellones auriculares de baja implantación con rotación posterior. El síndrome de Kabuki se caracteriza por sus anomalías faciales peculiares que se consideran son la única manifestación que puede orientar al diagnóstico del mismo sin excepciones. Recientemente se han identificado mutaciones sin sentido y de corrimiento del marco de lectura, entre otras en el gen MLL2 en aproximadamente el 75 % de los casos y en una menor proporción deleciones y mutaciones sin sentido en el gen KDM6A.
Palabras clave: síndrome Kabuki, anomalías cráneofaciales, MLL2, KDM6A, Colombia.
Abstract
Kabuki syndrome is a rare disease described by Kuroki and Niikawa in Japanese population in 1981. There are over 400 cases over the world and 5 cases described in Colombian population. Therefore this is the 6th Kabuki syndrome found in Colombia. We report a 2 years old female with Kabuki syndrome phenotype. Clinical examination showed: long palpebral fissures with eversion of the lateral third of the lower eyelids, a broad and depressed nasal tip, left palate and low setup ears. Kabuki syndrome includes facial features whit specific characteristics enough to classify the patients. However, there are some mutations in MLL2 gene present in almost 75 % of Kabuki syndrome. In addition there are some deletion and duplications abnormalities in KMD6A gene described in Kabuki syndrome patients.
Keywords: Kabuki, craniofacial anomalies, MLL2, KDM6A, Colombia.
INTRODUCCIÓN
El síndrome de Kabuki (SK) (OMIM 147929, OMIM 300867) es denominado así por la similitud entre la dismorfia facial que exhiben los pacientes con esta condición y las máscaras japonesas utilizadas en el teatro Kabuki. Es conocido también como "Síndrome Kabuki Makeup" y "Síndrome Niikawa Kuroki".
Los primeros reportes del SK fueron realizados por los investigadores japoneses Yoshikazu Kuroki y Norio Niikawa en 1981 (1-3).
Inicialmente se creyó que esta patología solo se encontraba en la etnia japonesa, con una incidencia de 1 en 32 000 nacidos vivos (3-4), pero a partir de la década de los noventa se han publicado más de 400 casos en diversos países, como Estados Unidos, India, México, Filipinas, China, Brasil, Vietnam, Arabia (4).
En cuanto a la frecuencia de esta patología según el sexo, la mayoría de reportes y revisiones no han encontrado discriminación entre hombres y mujeres; otros estudios muestran que esta patología se presenta en una relación hombre a mujer 1,6:1, respectivamente (4,5).
El síndrome de Kabuki se caracteriza por marcadas anomalías craneofaciales y físicas.
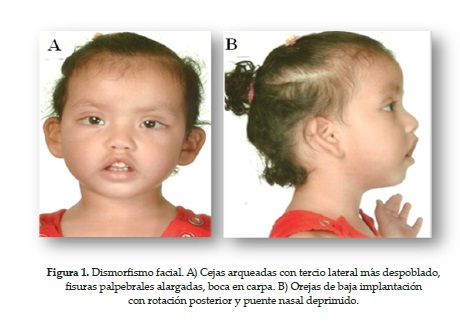
En 1988 Nikawaa et al. describieron las manifestaciones cardinales en las que basaban el diagnóstico de este síndrome, entre las que se destacan principalmente las características faciales peculiares, que incluyen fisuras palpebrales largas, eversión del párpado inferior, cejas arqueadas con tercio lateral despoblado, puente nasal ancho y/o deprimido, pabellones auriculares grandes o malformados, paladar alto o hendido La talla baja y el retraso mental son otras de las características mayor mente descritas; esta última ha sido encontrada en más del 90 % de los casos con coeficientes intelectuales de 89 o menos (6-9).
Con el tiempo se ha demostrado que las características faciales son la única manifestación que puede ayudar en el diagnóstico de síndrome de Kabuki sin excepciones, y la persistencia de las almohadillas en el pulpejo de los dedos y el acortamiento del quinto dedo de las manos deben considerarse igualmente manifestaciones características del síndrome.
En menor frecuencia se han descrito anomalías como micrognatia, ptosis, hipoacusia, escoliosis, dentición anormal, hipotonía, defectos cardiacos, renales, vértebras deformadas, estrabismo, hirsutismo, criptorquidia, deficiencia de la hormona del crecimiento, implantación del cabello baja, fosita o apéndice preauricular, entre otras (6-15).
La etiología de este síndrome es heterogénea. Diversas alteraciones cromosómicas estructurales se han descrito en pacientes con SK. Recientemente se identificaron mutaciones en el gen Mixed Lineage Leukimia 2 (MLL2) en un porcentaje entre 56-75 % de los pacientes con SK16-19, y en una menor proporción en el gen Lysine (K)-specific demethylase 6A (KDM6A) (20,21). Sería entonces de gran utilidad realizar pruebas moleculares en ambos genes, el MLL2 y el KDM6A, con el fin de correlacionar las características genotípicas y fenotípicas de los pacientes con sospecha de SK.
CASO CLÍNICO
Paciente femenina de 2 años y 6 meses, remitida a consulta genética por retraso psicomotor, paladar hendido y rasgos dismórficos. Producto de primera gestación, nacida por cesárea a las 37 semanas de gestación, con peso y talla adecuados para la edad.
Fue hospitalizada en la unidad de cuidados intensivos neonatales por depresión respiratoria, relacionada a aspiración de meconio, e hipertonía.
Desarrollo psicomotor: sostén cefálico: 3-4 meses; sedestación 6 meses; gateo 8 meses; se levantó con apoyo a los 12 meses; primeras palabras: 11 meses; con retraso en el desarrollo del lenguaje. Antecedente familiar de autismo en primo paterno.
Hallazgos positivos encontrados en el examen físico:
1. Fisuras palpebrales elongadas.
2. Eversión del tercio lateral del párpado inferior.
3. Telecanto.
4. Cejas arqueadas con tercio lateral más despoblado.
5. Pabellón auricular de baja implantación con rotación posterior.
6. Paladar blando hendido.
7. Boca en carpa.
8. Puente nasal deprimido.
9. Extremidades: adelgazamiento distal de las falanges y persistencia de almohadilla en el pulpejo de los dedos
Estos datos nos permiten clasificar a la paciente dentro de este síndrome.
Se le ordenó un cariotipo de bandas G, el cual fue normal (46, XX), y una resonancia magnética, en la que se encontró moderado retraso en la mielinización.
DISCUSIÓN
EL SK se caracteriza por anomalías faciales que se pueden identificar de forma temprana y permiten realizar el diagnóstico de este síndrome, el cual es básicamente clínico.
Cabe resaltar que en los últimos años se han hecho importantes avances en cuanto a estudios moleculares de esta enfermedad, con lo cual se ha logrado facilitar el diagnóstico preciso y el entendimiento fisiopatológico de este síndrome.
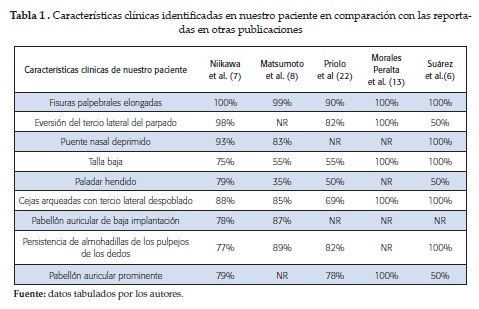
Las principales manifestaciones halladas en nuestra paciente se correlacionan con las publicadas por autores internacionales y nacionales, como puede apreciarse en la tabla 1. Encontramos que las características más comunes del SK son las fisuras palpebrales elongadas, la talla baja, la eversión del tercio lateral del parpado y las cejas arqueadas con tercio lateral despoblado.
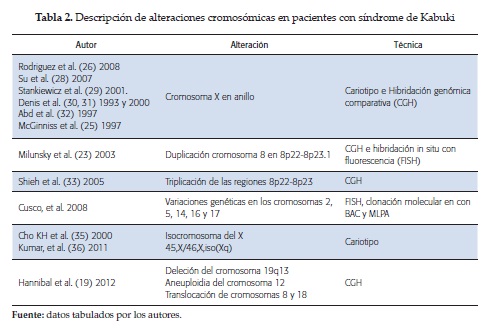
Es recomendable realizar inicialmente en pacientes con sospecha de SK un cariotipo, con el fin de determinar la presencia de anomalías cromosómicas estructurales, las cuales han sido descritas en algunos pacientes: duplicaciones en 8p22/8p23.1 (23-24), isocromosomas, cromosomas X en anillo25,-26; tambiéndescritas en pacientes con rasgos de síndrome de Turner, que han sido publicados como casos de síndrome de Turner-Kabuk (20, 27).
En caso de no poder evidenciar una alteración citogenética, los estudios moleculares son altamente recomendables por la base heterogénea de este síndrome (ver tabla 2).
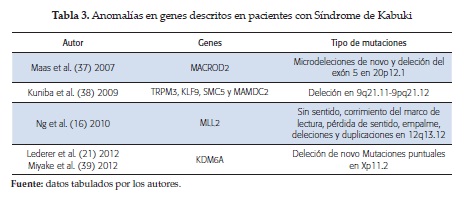
Las pruebas moleculares han identificado mutaciones en diversos genes, como el MACRO domain 2 (MACROD2), transient receptor potential catión channel C3 (TRPM3), kruppel like factor 9 (KLF9), structural maintenanace of chromosomes 5 (SMC5) y MAM domain containing 2 (MAMDC2), descritos en la tabla 3 (3, 7).
Los hallazgos de estos estudios no han sido replicados en otros pacientes. Sin embargo, el hallazgo molecular más significativo hasta el momento, referente al síndrome de Kabuki ha sidola identificación de los genes MLL2 y KDM6A.
Mutación en MLL2
La mutación en el gen MLL2 ha sido identificada en pacientes con síndrome de Kabuki por medio de análisis de la secuencia del exoma. Diferentes tipos de mutaciones en el gen MLL2 se han descrito en un 55 al 80 % de los casos, y las más comunes son las mutaciones sin sentido y de corrimiento del marco de lectura (figura 2 y tabla 4) (16-19, 23,40).
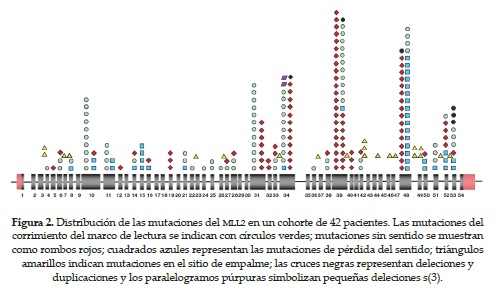

A pesar de que la heterogeneidad fenotípica de pacientes con SK es muy variable, se ha descubierto que la gran mayoría de pacientes con la típica dismorfia facial de este síndrome tienen una mutación patológica en el gen MLL240.
La presencia de mutaciones en este gen aumenta también la probabilidad de sufrir problemas nutricionales, anomalías renales, dislocaciones articulares y malformaciones palatinas (17, 40).
El gen MLL2 está situado en el brazo largo del cromosoma 12 (12q13.12), más específicamente, entre los pares de base 49.412.757 a la 49.449.106, y codifica la enzima Histona-lisina N-metil-transferasa, de 5.537 aminoácidos. Esta enzima hace parte del grupo de proteínas Tritorax, que son un grupo heterogéneo de proteínas cuya función principal es regular y mantener la expresión genética; la MLL2 específicamente se subclasifica como modificadora de histona en el grupo Tritorax41-42.
La función de la enzima Histona-lisina N-metil-transferasa es transferir de uno a tres grupos metilo desde el cofactor S-Adenosil metionina a los residuos de lisina y arginina de la histona H3 (H3K4me), que representa un marcador específico de activación transcripcional cumpliendo de esta manera con un papel muy importante en la modificación y regulación epigenética de las células (17,43). Esta enzima hace parte de un largo complejo proteínico llamado ASCOM, que ha sido reconocido como regulador transcripcional de la beta-globina y de genes que codifican para receptores de estrógeno (44-45).
Mutación en KDM6A
El gen KDM6A contiene 29 exones y está localizado en el cromosoma X, más específicamente, en Xp11.2, entre los pares de base 44,732,422 y 44,971,84646. El KDM6A es uno de los genes que escapa a la inactivación del cromosoma X y codifica una proteína de 1,401 aminoácidos, la cual está compuesta por dos dominios funcionales. Un dominio catalítico se encarga de catalizar la desmetilación del H3K27; esta desmetilación media la expresión de varios genes relacionados con el ciclo celular (21). El otro dominio del KDM6A juega un rol importante en la remodelación de la cromatina al interactuar con el complejo SWI/SNF (switch/sucrose nonfermentable), que contiene el factor activador de transcripción Brg121. Por último, el KDM6A interactúa con la familia de genes T-Box, los cuales actúan en el mesodermo en la formación del corazón y las vértebras (21).
El MLL2 junto con el KDM6A juegan un papel importante en el control epigenetico, al encargarse de la activación de la cromatina por contra regulación del grupo de proteínas polycomb (PcG), las cuales han sido relacionadas con procesos patológicos como el cáncer, a través de la desregulación a nivel epigenético de genes supresores de tumores como BRCA1, p16 y p53, entre otros (47). Estos además actúan en la regulación de genes específicos de músculo durante la embriogénesis y el desarrollo.
En enero de 2012 Lederer et al. (21) reportaron 22 casos de pacientes con SK que resultaron negativos para mutaciones en el gen MLL2, pero 3 de ellos presentaron deleciones de novo en el gen KDM6A. En esta investigación se demostró también la relación entre el cromosoma X y el SK, que había sido sugerido previamente se logró entonces evidenciar que el SK es un síndrome genéticamente heterogéneo. Posteriormente Miyake et al. (20) en octubre del mismo año describieron 3 casos de SK con mutaciones puntuales del gen KDM6A, 2 mutaciones sin sentido y una deleción de tres pares de base en un total de 32 pacientes evaluados.
La identificación del KDM6A en la patogénesis del SK aumenta el papel de los factores de discapacidad intelectual y malformaciones congénitas (21).
Con estos hallazgos se manifiesta la necesidad de secuenciar el gen KDM6A, como ayuda diagnostica en casos de sospecha de SK, sobre todo en pacientes con estudios del gen MLL2 negativos, a pesar de ser poco frecuente (3).
Otros genes
Mutaciones del MACROD2: Maas et al. (37) en 2007 describieron el caso de un paciente con SK que presentaba una microdeleción de novo en el exon 5 de este gen, el cual está ubicado en el cromosoma 20p12.1, contiene 17 exones y tiene una extensión de 2.06 Mb, localizado entre los pares de base 13.92-15.98. Su función no es conocida (37).
Mutaciones de TRPM3, KLF9, SMC5, MAMDC2: Kuniba et al. (38) en 2009 publicaron el estudio del cariotipo molecular en 17 pacientes y la detección de mutaciones en 41 pacientes con síndrome de Kabuki, en los que encontraron 7 alteraciones en el número de copias, tres regiones suprimidas y cuatro regiones duplicadas entre los pacientes.
Entre los siete loci solo la región de 9q21.11-q21.12 participa en la codificación de estos 4 genes; en los cuales, si bien no se demostró ser la causa del SK, sí podrían tener relación con las manifestaciones que presentaban estos pacientes.
El gen TRPM3 el cual es un mediador para la entrada de iones de calcio por medio de sus canales, el cual ha sido relacionado con casos de paladar hendido (38).
Síndrome Turner-Kabuki
La herencia ligada al cromosoma X también se ha descrito en la génesis del SK. Anomalías de los cromosomas sexuales han sido reportadas en varias ocasiones y algunos de los hallazgos son compartidos con el síndrome de Turner (ST) (3,20).
En 1994 se reportó el caso de una niña de 2 años, a quien se le diagnosticó por análisis citogenético la presencia de ST. Esta paciente presentaba, además del fenotipo característico de ST, rasgos faciales dismórficos y microcefalia, por lo cual se denominó como síndrome Turner-Kabuki.
A partir de este hallazgo se empezaron a publicar diferentes estudios que sugirieron la posible relación entre estos dos síndromes (4, 27).
Los hallazgos más significativos hasta el momento que asocian genéticamente al ST y al SK son los cromosomas X en anillo, el isocromosoma y la trisomía X (4).
CONCLUSIÓN
El síndrome de Kabuki es una rara entidad genética con hallazgos faciales peculiares que orientan para realizar el diagnóstico clínico. Entre los hallazgos fenotípicos se incluyen fisuras palpebrales largas, eversión del párpado inferior, cejas arqueadas con tercio lateral despoblado, puente nasal ancho y/o deprimido y pabellones auriculares grandes o malformados.
Su etiología es heterogénea, y se han descrito anomalías cromosómicas o génicas, por lo cual se recomienda el estudio citogenético y molecular para poder orientar el asesoramiento genético en el núcleo familiar. El caso presentado es el sexto reportado a nivel de Colombia y estamos a la espera de la realización del estudio de ADN para dilucidar la etiología molecular de la paciente estudiada. Finalmente, se realizó una revisión y actualización de la literatura relacionada con el conocimiento clínico y molecular del síndrome de Kabuki.
Conflicto de intereses: Los autores declaran que no hay conflicto de intereses en este manuscrito.
Financiación: Universidad del Norte.
REFERENCIAS
1. Niikawa N, Matsuura N, Fukushima Y, Ohsaw T, Kajii T. Kabuki make-up syndrome: A Syndrome of mental retardation, unusual facies, large and protuding ears, and postnactal growth deficiency. J Pediatr 1981; 99:565-569. [ Links ]
2. Kuroki Y, Suzuki Y, Chio H, Hata A, Matsui I. A new malformation; Syndrome of long palpebral fissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal dwarfism and mental retardation. J Pediatr 1981; 99:570-573. [ Links ]
3. Bögershausen N, Wollnik B. Unmasking Kabuki syndrome. Clin Genet 2012;83(3):201-11. Doi: 10.1111/cge.12051 [ Links ]
4. Yemisi Bokinni. Kabuki syndrome. Journal of Human Genetics 2012;57:223-227. [ Links ]
5. Kasdon BD, Fox JE. Kabuki syndrome: diagnostic and treatment considerations. Mental Health in Family Medicine 2012; 9(3):171-9. [ Links ]
6. Suarez JL et al. Síndrome de Kabuki. An Pediatr (Barc). 2012;77(1):51-6. Doi:10.1016/j.an pedi. 2012.01.016 [ Links ]
7. Niikawa N, Kuroki Y, Kajii T, Matsuura N, Ishikiriyama S, Tonoki H, y cols. Kabuki make-up (Niikawa-Kuroki) syndrome: a study of 62 patients. Am J Med Genet 1988; 31: 565-90. [ Links ]
8. Matsumoto N, Niikawa N. Kabuki make-up syndrome: a review. Am J Med Genet 2003; 117: 57-65. [ Links ]
9. Guerrero JL, Contreras GA. Síndrome de Kabuki: caracterización clínica, estudios genéticos, manejo preventivo de las complicaciones y asesoría genética. méd.uis. 2012;25(1):19-27. [ Links ]
10. Serbati N, Nassereddine S, Dehbi H, and Nadifi S. Clinical Heterogeneity of Kabuki Syndrome: Study of Moroccan Patients. International Journal of Life Sciences Biotechnology and Pharma Research 2012: 1(1):68-74. [ Links ]
11. Aviña-Fierro JA, Pérez N. Síndrome de Kabuki, Informe de un caso interesante. Acta Pediatr Mex 2006; 27(6), 349-51. [ Links ]
12. Pascual-Castroviejo I, Pascual-Pascual SI, Velázquez-Fragua R, Palencia R. Síndrome del maquillaje Kabuki. A propósito de 18 casos españoles. Rev Neurol 2005; 40(8): 473-8. [ Links ]
13. Chiong-Quesada C, Morales-Peralta E. Síndrome Kabuki. Presentación de dos casos. Revista Cubana de genética comunitaria 2008; 2(2):73-76. [ Links ]
14. González C, García-Alix A, del Campo M, Garrido J, Quero J. Síndrome de Kabuki, un cuadro reconocible desde la infancia precoz. An Esp Pediatr 1997;47:429-431. [ Links ]
15. S Ohdo, H Madokoro, T Sonoda, y cols. Kabuki make-up syndrome (Niikawa-Kuroki syndrome) associated with congenital heart disease. Journal of Medical Genetics 1985; 22: 126-127. [ Links ]
16. Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, y cols. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet 2010 September; 42(9): 790-793. Doi:10.1038/ng.646. [ Links ]
17. Micale y cols. Mutation spectrum of MLL2 in a cohort of kabuki syndrome patients; Orphanet Journal of Rare Diseases 2011, 6:38. http://www.ojrd.com/content/6/1/38 [ Links ]
18. Aimée D.C. Paulussen y cols. MLL2 Mutation Spectrum in 45 Patients with Kabuki Syndrome; Human Mutation. Mutation in Brief [en línea] 2010; 32: E2018-E2025. [ Links ]
19. Hannibal MC, Buckingham KJ, Ng SB et al. Spectrum of MLL2 (ALR) mutations in 110 cases of Kabuki syndrome. Am J Med Genet A 2011: 155A: 1511-1516. doi:10.1002/ajmg.a.34074. [ Links ]
20. Miyake N et al. KDM6A Point Mutations Cause Kabuki Syndrome. Human mutation 2013; 34(1): 108-110. [ Links ]
21. Lederer et al. Deletion of KDM6A, a Histone Demethylase Interacting with MLL2, in Three Patients with Kabuki Syndrome. The American Journal of Human Genetics 2012 Jan;90: 119-124, 13. [ Links ]
22. Priolo M, Micale L, Augello B, Fusco C, Zucchetti F, Prontera P. Absence of deletion and duplication of MLL2 and KDM6A genes in a large cohort of patients with Kabuki syndrome. Molecular Genetics and metabolism 2012; 107: 627-629. [ Links ]
23. Milunsky JM, Huang XL. Unmasking Kabuki syndrome: chromosome 8p22-8p23.1 duplication revealed by comparative genomic hybridization and BAC-FISH. Clin Genet 2003: 64: 509-516. [ Links ]
24. Kendra W Kimberley, Colleen A Morris and Holly H Hobart. BAC-FISH refutes report of an 8p22-8p23.1 inversion or duplication in 8 patients with Kabuki syndrome. BMC Medical Genetics 2006, 7:46 Doi:10.1186/1471-2350-7-46 [ Links ]
25. McGinniss et al. Ring Chromosome X in a Child With Manifestations of Kabuki Syndrome. American Journal of Medical Genetics 1997;70:37-42. [ Links ]
26. Rodriguez et al. A Small and Active Ring X Chromosome in a Female With Features of Kabuki Syndrome. American Journal of Medical Genetics 2008; Part A 146A:2816-2821. [ Links ]
27. Wellesley DG, Slaney S. Kabuki make-up and Turner syndromes in the same patient. Clin Dysmorphol. 1994 Oct; 3(4):297-300. [ Links ]
28. Su PH, Kuo PL, Chen SJ et al. Kabuki make-up (Niikawa-Kuroki) syndrome with mosaicism ring chromosome X and incomplete XIST gene expression. Acta Paediatr Taiwan 2007; 48: 28-31. [ Links ]
29. Stankiewicz P, Thiele H, Giannakudis I et al. Kabuki syndrome-like features associated with a small ring chromosome X and XIST gene expression. Am J Med Genet 2001; 102: 286-292. [ Links ]
30. Dennis N, Coppin B, Turner C, Skuse D, Jacobs P. A clinical, cytogenetic and molecular study of 47 females with r(X) chromosomes. Ann Hum Genet 2000; 64: 277-293. [ Links ]
31. Dennis NR, Collins AL, Crolla JA, Cockwell AE, Fisher AM, Jacobs PA. Three patients with ring (X) chromosomes and a severe phenotype. J Med Genet 1993; 30: 482-486. [ Links ]
32. Abd SE, Wilson L, Howlin P, Patton MA, Wintgens AM, Wilson R. Agenesis of the corpus callosum in Turner syndrome with ring X. Dev Med Child Neurol 1997; 39: 119-124. [ Links ]
33. Shieh JT, Hudgins L, Cherry AM, Shen Z, Hoyme HE. Triplication of 8p22-8p23 in a patient with features similar to Kabuki syndrome. Am J Med Genet A 2006; 140: 170-173. [ Links ]
34. Cusco I, del Campo M, Vilardell M et al. Array-CGH in patients with Kabuki-like phenotype: identification of two patients with complex rearrangements including 2q37 deletions and no other recurrent aberration. BMC Med Genet 2008; 9: 27. [ Links ]
35. Cho KH et al. Two Cases of Kabuki Make-up Syndrome Including One Case Associated with Xq Isochromosome. Korean J Pediatr 2000 August;43(8):1111-1115. [ Links ]
36. Kumar JM, Gowrishankar K, Vasanthi T, Ashok Kumar R, Jayasudha T. Isochromosome X mosaicism in a child with Kabuki syndrome phenotype: a rare cytogenetic association. Indian J Hum Genet 2011; 17: 241-243. [ Links ]
37. Maas NM, Van de Putte T, Melotte C et al. The C20orf133 gene is disrupted in a patient with Kabuki syndrome. J Med Genet 2007; 44: 562-569. [ Links ]
38. Kuniba H, Yoshiura K, Kondoh T et al. Molecular karyotyping in 17 patients and mutation screening in 41 patients with Kabuki syndrome. J Hum Genet 2009; 54: 304-309. [ Links ]
39. Miyake et al. KDM6A point mutations cause kabuki syndrome. Human mutation 2013; 34(1): 108-110. [ Links ]
40. Banka S et al.: How genetically heterogeneous is Kabuki syndrome?: MLL2 testing in 116 patients, review and analyses of mutation and phenotypic spectrum. Eur J Hum Genet 2012 Apr;20(4):381-8. Doi: 10.1038/ejhg.2011.220. Epub 2011 Nov 30. [ Links ]
41. Schuettengruber B, Chourrout D, Vervoort M, Leblanc B, Cavalli G. Genome regulation by polycomb and trithorax proteins. Cell. 2007 Feb 23;128(4):735-45. [ Links ]
42. Lan F, Bayliss PE, Rinn JL, Whetstine JR, Wang JK, Chen S, Iwase S, Alpatov R, Issaeva I, Canaani E, Roberts TM, Chang HY, Shi Y. A histone H3 lysine 27 demethylase regulates animal posterior development. Nature 2007 Oct 11;449(7163):689-94. Epub 2007 Sep 12. [ Links ]
43. Schuettengruber B, Martinez AM, Iovino N, Cavalli G. Trithorax group proteins: switching genes on and keeping them active. Nat Rev Mol Cell Biol 2011 Nov 23;12(12):799-814. [ Links ]
44. Rigen Mo, Sambasiva M. Rao, Yi-Jun Zhu. Identification of the MLL2 Complex as a Coactivator for Estrogen Receptor. JBC Papers in Press 2006 April 7. [ Links ]
45. Issaeva I, Zonis Y, Rozovskaia T, Orlovsky K, Croce CM, Nakamura T, Mazo A, Eisenbach L, Canaani E. Knockdown of ALR (MLL2) Reveals ALR Target Genes and Leads to Alterations in Cell Adhesion and Growth. Mol. Cell. Biol. 2007; 27(5):1889. [ Links ]
46. Mansour AA et al. The H3K27 demethylase Utx regulates somatic and germ cell epigenetic reprogramming. Nature 2012 Aug 16;488(7411):409-13. Doi: 10.1038/nature11272. [ Links ]
47. Davalos Mercedes y Recillas felix. La vía RB/E2F y la familia de las proteínas represoras Polycomb en el desarrollo de cáncer. Revista especializada en ciencias Quimico-Biológicas 2011;14(1):38-50. [ Links ]

