











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCCIÓN
El síndrome de Townes-Brocks [TBS (OMIM 107480] o síndrome REAR (Renal, Ear, Anal y Radial) fue descrito inicialmente en 1972 1. Se caracteriza por la triada consistente en ano imperforado (84 %), orejas displásicas, que incluyen desde apéndices preauriculares hasta alteración auditiva neurosensorial y/o conductiva (87 %), y malformaciones en pulgares, entre las cuales se han descrito pulgares trifalángicos, polidactilia e hipoplasia (89 %) 1 Así mismo, se han documentado alteraciones renales (ectopia, hipoplasia, riñones poliquísticos y reflujo vesicoureteral) que pueden conducir a enfermedad renal crónica (42 %), cardiopatías congénitas (25%), malformaciones en extremidades inferiores (52 %) y malformaciones genitourinarias (36 %) 1. El diagnóstico es clínico y se relaciona con mutaciones en el gen SALL1 o factor de transcripción Sal-like protein 1, mapeado en el cromosoma 16q12.1 1. Sin embargo, alteraciones cromosómicas como translocaciones e inversiones pericéntricas y deleciones citogenéticas que comprometen 16q13 han sido registradas 1. Su modelo de herencia es autosómica dominante y en el 50% es secundario a mutaciones de novo1.
En este trabajo se presenta el caso de un recién nacido con malformaciones compatibles con TBS y la revisión del estado del arte de esta patología.
CASO CLÍNICO
Recién nacido masculino, producto de segundo embarazo, madre de 23 años, sin exposición a teratógenos y/o a agentes mutagénicos durante la gestación. La madre cursó con diabetes gestacional manejada con dieta. El perfil infeccioso para hepatitis, VIH, sífilis, toxoplasmosis, rubeola, citomegalovirus y herpes simple fue negativo. Ecografía obstétrica a las 35 semanas reportó polihidramnios, agenesia renal izquierda, mesomelia de extremidades superiores con agenesia de radio bilateral. Parto por cesárea a las 37 semanas, observándose un neonato hipotónico, con mala adaptación neonatal, APGAR 2-3/10 y 4/20, al minuto y 5 minutos de vida, respectivamente, por lo cual se iniciaron maniobras de reanimación. Falleció posteriormente por paro cardiorrespiratorio. Las medidas antropométricas fueron: peso 3060 gr (P25, -0.66 z), talla 49 cm (P14, -1.06 z), perímetro cefálico 34 cm (P18, -0.9 z), perímetro torácico 31 cm, perímetro abdominal 28 cm.
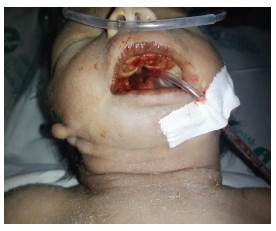
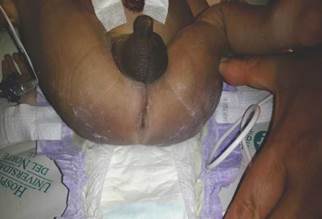
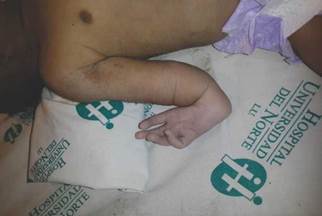
Al examen físico se evidenció ausencia de pabellón auricular bilateral, apéndices cutáneos en mejillas, paladar hendido completo, glosoptosis, micrognatia, coanas no permeables, cuello corto (figuras 1 y 2). Se auscultó soplo sistólico grado II/VI con bradicardia. Se evidenció ano imperforado, rafe perineal prominente (figura 3); mesomelia en extremidades superiores y agenesia de pulgares (figura 4). Estudio de imágenes mostraron infiltrado de tipo vidrio esmerilado en ambos campos pulmonares, cardiomegalia global, intestino dilatado con aumento de contenido gaseoso, curva escoliótica lumbar de convexidad izquierda, dos hemivértebras en L2 y L5, ausencia bilateral de radios y pulgares (figura 5), y agenesia de riñón izquierdo. Estudio citogenético mostró un cariotipo 46, XY normal. No fue posible la realización de estudios moleculares, sin embargo, con base en los hallazgos clínicos se clasifica como un síndrome de Townes-Brocks.


DISCUSIÓN
Historia
El TBS fue descrito por primera vez en 1972 por Phillip Townes y Eric Brocks 2 al observar un padre y cinco de sus siete hijos con ano imperforado, pulgares trifalángicos, fusión de metatarsos, pulgares supernumerarios, pulgares bífidos, sordera neurosensorial y deformidades en orejas. En 1978 se estableció como herencia autosómica dominante en individuos con anomalías anales, radiales, auditivas y renales, incluyendo hipoplasia y displasia renal, por lo que otorgaron el acrónimo REAR (Renal, Ear, Anal y Radial) 3. En 1984 se reportó el primer caso de un paciente TBS y cardiopatía congénita asociada 4. En 1987 se describió por primera vez la expresividad variable de TBS en familiares de primer grado de consanguinidad 5. En 1993 se establecieron las características principales de la entidad 6 y se corroboró la variabilidad clínica de TBS 7.
Epidemiología
Desde su descripción inicial hasta mediados de 2010 se reportaron 66 casos diagnosticados por criterio clínico y/o pruebas de laboratorio. La prevalencia de la patología es de 1 en 250 000 personas 8, debido a que con frecuencia el diagnóstico se complica por la variedad de signos y síntomas compartidos con otras entidades, enmascarando su prevalencia real.
Teniendo en cuenta que es un síndrome de difícil diagnóstico, son pocos los estudios epidemiológicos alrededor del mundo que establecen una prevalencia para esta entidad. En Colombia solo existen estudios que establecen la prevalencia de anomalías craneofaciales congénitas de forma aislada y no sindromática, en presencia de factores de riesgo como la diabetes gestacional 9-12, siendo las anomalías de oreja y hendiduras faciales las más frecuentes, y el ano imperforado y la agenesia renal unilateral las de menor tasa.
Genética
El gen SALL1, localizado en 16q12.1, codifica para la proteína de su mismo nombre, el factor de transcripción Sal-like protein 1, y contiene dominios de zinc, glutamina, prolina, alanina y serina 6. El gen ocupa 14.1 kb desde el codón de inicio al de parada y contiene 4 exones codificantes y dos intrones. La proteína se encuentra en el núcleo celular, se une a la heterocromatina y contiene dominios represores en N-terminal y región central 1. El gen se expresa en humanos durante el desarrollo cerebral del feto, lo cual sugiere que su déficit puede justificar el retraso mental de algunos pacientes afectados.
En la búsqueda de la etiología genética de esta patología se describieron inicialmente anomalías cromosómicas como inversión pericéntrica del cromosoma 16 con puntos de ruptura en p11.2 y q12.1 7 y translocaciones entre los cromosomas 5 y 16 [46,XX,t(5;16)(p15.3;q12.1)] 6 y entre los cromosomas 2, 11 y 16 13. Así mismo, se describieron deleciones citogenéticas en el brazo largo del cromosoma 16, que incluyeron la porción distal de 16q12.1 y se extendieron hasta 16q13 14. En el cariotipo del caso presentado no se evidenció alguna alteración cromosómica.
Al combinar las regiones implicadas reportadas, se estableció que la región susceptible a mutaciones causante de TBS se encuentra a 1-1.2 Mb distal a la banda 16q12.1 15. Debido a lo anterior y si la clínica no es concluyente, la identificación de una variante patológica de SALL1 establecerá el diagnóstico.
La mutaciones más comunes son mutaciones sin sentido: c.826C>T (p.Arg276Ter) y c.3160C>T (p.Arg1054Ter), asociándose la primera a defectos cardiacos y neurológicos más severos. Se han encontrado otros tipos de mutación en el gen SALL1 asociados a expresividad variable 16. Por ejemplo, a pesar de que las alteraciones endocrinas raramente se asocian a mutación de SALL116-18, se ha descrito un paciente con TBS mutación de corrimiento del marco de lectura (c.3414_ 3415delAT) y deficiencia de hormona de crecimiento 19.
Hasta el 2010 se reportaron 42 mutaciones sin sentido, 104 mutaciones por desplazamiento del marco de lectura y 8 mutaciones por deleciones 20. Existen 29 diferentes polimorfismos no patológicos 21, mientras que todas las variantes patológicas se localizan en el exón 2 e intrón 2 22. De las 56 variantes patológicas, 46 han presentado su hotspot en el exón 2, de 802 pb de tamaño. No se han realizado correlaciones genotipo-fenotipo para la mayoría de las variantes patogénicas.
El punto crítico de la patogénesis es la cantidad funcional de proteína SALL1 que explica la variabilidad fenotípica en TBS. La deleción de un alelo genera reducción del 50 % de la funcionalidad de la proteína (función residual), referido como haploinsuficiencia de SALL1, lo cual fue detectado inicialmente por una deleción de 75 kb de toda la región codificante en tres familias que generó fenotipos leves 20,23.
Existen dos pruebas moleculares para confirmar TBS. En primera instancia se encuentra el análisis de secuencia de la región codificante de SALL1 desde el exón 1 al 3 y la búsqueda de deleción o duplicación si no se encuentra la variante patogénica principal. Con ambos métodos implementados se identifica la mutación en el 70 % de los casos de los individuos con la triada característica 1.
El análisis de un panel multigen se recomienda en segunda instancia cuando un paciente con manifestaciones clínicas de TBS presenta pruebas negativas para el gen SALL1, por lo que se deberán buscar variantes patológicas de SALL4 (20q13.2) 24. Adicionalmente se debe considerar realizar pruebas moleculares de SALL4 si hay presencia de manifestaciones radiales, anomalía de Duane o síndrome de Okihiro 25. Puesto que en el caso presentado se evidenció agenesia radial y manifestaciones atípicas, era necesario realizar panel multigen.
Diagnóstico clínico
El diagnóstico de TBS o REAR se establece con la presencia de las tres características clínicas mayores: ano imperforado o estenosis anal, orejas displásicas y malformaciones en pulgar sin hipoplasia de radio. Si solo dos características mayores se encuentran en el individuo, la sospecha diagnóstica será apoyada con la presencia de hallazgos clínicos menores, como la alteración neurosensorial y/o de la audición conductiva, malformaciones de pie, malformación genitourinaria, cardiopatía congénita, entre otros. Las características atípicas, como hipoplasia de radio, labio y paladar hendido, deben ser consideradas inicialmente no sugestivas de TBS, aunque se presenten en menor proporción 1. A pesar de que el recién nacido presentó dos características mayores (ano imperforado y orejas displásicas) y varias menores, generando el diagnóstico clínico, también se evidenciaron manifestaciones clínicas atípicas como agenesia de pulgar y radio y paladar hendido.
Dentro de las anomalías auriculares y de audición se incluye el plegamiento del hélix superior y un antihélix pequeño, en ocasiones con presencia de fosas y apéndices auriculares, presenciados en el caso reportado. La hipoacusia es común en TBS, con un rango variable de severidad, siendo usualmente congénitas y principalmente neurosensorial 13,26.
Los defectos más comunes de las extremidades son pulgar trifalángico y polidactilia preaxial con un dígito bien formado y uno vestigial. También son comunes la bifurcación, desviación ulnar y la apariencia ancha de la falange distal del pulgar. La hipoplasia del pulgar es frecuente, sin embargo, su asociación con anomalías del radio, como es la hipoplasia, pertenecen al grupo de características atípicas de TBS, las cuales en presencia de ano imperforado hacen sospechar la asociación VACTERL principalmente 4,13. El caso reportado se consideró que la agenesia bilateral de radios y pulgares eran manifestaciones clínicas atípicas de TBS.
La anomalía anal más comúnmente reportada es el ano imperforado, el cual varía desde un orificio cubierto por piel hasta grados de mayor severidad. Usualmente se encuentran fistulas recto-vaginales o rectouretrales, estenosis anal, exceso de piel perianal, estreñimiento crónico y reflujo gastroesofágico 15,27. Igualmente, se han descrito múltiples anomalías genitourinarias, como la displasia o hipoplasia de riñón uni- o bilateral, agenesia renal, riñones poliquísticos, valvas uretrales posteriores, reflujo vesicoureteral y estenosis meatal. Así mismo, se puede presentar falla renal, rafe perineal prominente, escroto bífido, criptorquidia, hipospadias y aplasia vaginal con útero bífido 15,28. Además del ano imperforado, el rafe perineal prominente y la agenesia renal unilateral del paciente no fue posible descartar otras anomalías genitourinarias por medio de estudios complementarios debido al fallecimiento temprano del mismo.
A pesar de que se han descrito cardiopatías congénitas en el 50 % de los individuos con la mutación más común (p.Arg276Ter) 29 y en el 25 % de personas con otras variantes de SALL117,30, la relación entre defectos cardíacos y TBS no ha sido comprobada. En nuestro reporte se encontró compromiso cardiaco clínico dado por la presencia de soplo sistólico grado II/VI y cardiomegalia global. No se realizó ecocardiograma por fallecimiento temprano del paciente.
Las manifestaciones oculares se consideran ocasionales, debido a su baja prevalencia. Se han descrito casos con coloboma de iris y coriorretiniano asociado a pérdida de visión, cataratas lamelares y microftalmia 31-33, la cual se evidenció de forma bilateral en el caso reportado.
No existe consenso sobre la presencia de anormalidades estructurales de la columna vertebral en TBS, puesto que la presencia de estas manifestaciones puede ser utilizada en ocasiones para realizar diagnóstico diferencial con la asociación VACTERL y/o síndromes con malformación anal y de manos 13,34. Sin embargo, se han evidenciado defectos leves esqueléticos en 9 % de los individuos con TBS 1, incluyendo anormalidades en arcos costales y articulaciones. Además, se han descrito tres casos de pacientes con TBS y escoliosis, que presentaban retraso mental 35,36. Una curva escoliótica lumbar de convexidad izquierda y dos hemivértebras en L2 y L5 fueron los hallazgos encontrados en el caso presentado
Diagnóstico diferencial
Las características de TBS se superponen con diversos síndromes y asociaciones (tabla 1), entre las cuales se encuentra la asociación VACTERL, caracterizada por la presencia de anomalías vertebrales, ano imperforado, cardiopatías, fistula traqueoesofágica, atresia esofágica, anomalías renales y/o radiales y defectos en extremidades. En TBS no se presenta fístula traqueoesofágica y es rara la presencia de anomalías vertebrales, mientras que en VACTERL no es típica la presencia de anomalías en pabellón auricular (apéndices preauriculares) y pérdida auditiva 37,38. Algunos pacientes con VACTERL presentan anemia de Fanconi, aplasia o hipoplasia de radio y pulgar, pulgares supernumerarios, pulgares bífidos y anomalías auditivas, que también pueden ser observadas en pacientes con TBS 39-42.
Tabla 1 Diagnósticos Diferenciales Sindromáticos

TBS: Síndrome de Townes-Brocks, VACTERL: Asociación VACTERL, BGS: Síndrome de Baller-Gerold, OAV: Oculo-Auriculo-Vertebral Spectrum o GS: Síndrome de Goldenhar, MCS: Síndrome de Moescher Clarren, OS: Síndrome de Okihiro, HOS: Síndrome de Holt-Oram, CHARGE: Síndrome CHARGE.
+ Manifestación clínica frecuente
+/- Manifestación clínica ocasional
Es necesario considerar el síndrome de Baller-Gerold (BGS) en segunda instancia, el cual cursa con craneosinostosis, alteraciones auriculares, micrognatia, hipoplasia o aplasia radial y de pulgar, defectos anales y urogenitales 43, de las cuales la mayoría se encuentran superpuestas con TBS. Sin embargo, una característica para diferenciarlos es la craneosinostosis que está presente en el 100 % de los pacientes con BGS 13,43, y que en el caso reportado no se evidenció.
En tercer lugar se encuentra el Espectro Oculoauriculovertebral (OAV) o síndrome de Goldenhar (GS), caracterizado por hipoplasia malar, maxilar o mandibular, macrostomia, alteraciones auditivas, hemivértebras o hipoplasia vertebral, malformaciones del sistema nervioso central, cardiopatías y anomalías genitourinarias 44. Así mismo, existe el síndrome de Moeschler Clarren (MCS), el cual es la presencia de OAV o GS asociado a defectos radiales como hipoplasia o agenesia de pulgar y/o radio, cardiopatías, anomalías renales y del sistema nervioso central 45. Para diferenciar OAV o GS y MCS de TBS, se debe tener en cuenta que en las dos primeras el ano imperforado no se considera característica típica u ocasional, el cual sí fue evidenciado en nuestro caso.
El síndrome de Okihiro (OS) se constituye por hipoplasia de radio y cúbito, hipoplasia o agenesia de pulgar, malformaciones renales y vertebrales junto con la presencia de la anomalía de Duane, que consiste en estrabismo congénito y retracción ocular en aducción 46, el cual es la diferencia principal con TBS. Si la clínica no es concluyente, se debe realizar un panel multigen, debido a que la mutación que causa OS se encuentra en SALL447.
En sexta instancia se encuentra el síndrome de Holt-Oram (HOS), el cual se caracteriza por malformaciones en extremidades superiores unilaterales y/o simétricas, como agenesia de pulgar, focomelia, hipoplasia o aplasia de radio, anomalía de carpos, malformaciones cardiacas congénitas y defectos de conducción cardiacos 48. Se diferencia de TBS en que no se ha documentado ano imperforado, malformaciones auditivas o renales.
El síndrome CHARGE es la séptima entidad que se caracteriza por coloboma de iris y retina, cardiopatía congénita, atresia de coanas, labio y paladar hendido, malformaciones genitales y auditivas 49. Sin embargo, es rara la presencia de ano imperforado y no se dan documentado malformaciones renales y de radio como ocurre en TBS.
Tratamiento
El tratamiento en TBS está enfocado en corregir las alteraciones anatómicas que comprometan la funcionalidad, por lo que es necesario un abordaje multidisciplinario priorizando aquellas manifestaciones que ponen en peligro la vida del paciente. Por ejemplo, en caso de una malformación anorrectal, no se debe realizar un tratamiento quirúrgico dentro de las primeras 24 horas de vida, debido a que se necesita de una presión intraluminal significativa para que el meconio pueda ser conducido. Dependiendo de la clínica del paciente con malformación anorrectal se puede realizar reparación primaria con anorrectoplastia sagital posterior, con o sin colostomía 50,51.
Igualmente, se debe evaluar la función renal periódicamente y realizar imágenes diagnósticas en la etapa neonatal para considerar la necesidad de terapia de remplazo renal, trasplante renal, nefrostomías o nefrectomía. Así mismo, las alteraciones auditivas son principalmente neurosensoriales con pequeños componentes conductivos, por lo cual la colocación de un implante coclear evita consecuencias en el neurodesarrollo.
Consejería genética
El TBS es heredado de forma autosómica dominante, con penetrancia completa, pero con expresividad y pronóstico altamente variable 52,53. Un 50 % de las personas con mutación de SALL1 tienen un padre afectado, mientras que el otro 50 % tienen la enfermedad como resultado de una mutación de novo, que ocurre en el 87.5 % en el cromosoma 16 paterno adquirido 21.
En el caso expuesto se les brindó consejería genética a los progenitores, y se le indicó que el síndrome es el resultado de una mutación de novo y el riesgo de recurrencia para futuros embarazos es bajo.
El diagnóstico prenatal se realiza determinando la presencia de la mutación SALL1 en el ADN extraído de células fetales por amniocentesis o vellosidades coriónicas entre las 15-18 o 10-12 semanas de gestación, respectivamente. Si no es posible realizar lo anterior, se debe evaluar el fenotipo del feto por ultrasonido de alta definición, el cual en el caso reportado evidenció inicialmente agenesia renal izquierda, mesomelia de extremidades superiores y agenesia de radio bilateral.

