










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Cardiología
Print version ISSN 0120-5633
Rev. Col. Cardiol. vol.11 no.8 Bogota Apr. 2005
Cardiomiopatía amiloidea
Cardiac amyloidosis
Jorge E. Marín, MD.; Mauricio Duque, MD.; Luis E. Medina, MD., William Uribe, MD.; Jorge E. Velásquez, MD.
Medellín, Colombia
Servicio de electrofisiología, arritmias y marcapasos. Cardiología, Clínica Medellín, Medellín. Colombia.
Correspondencia: Mauricio Duque, MD. Cardiología. Clínica Medellín. Calle 53 No. 46-38, segundo piso, Medellín, Colombia.
La amiloidosis es una enfermedad infiltrativa sistémica que compromete al corazón. Tiene orígenes genéticos y es una causa importante de cardiomiopatía restrictiva. Puede comprometer todas las estructuras cardiacas con predilección por el tejido miocárdico. Su manifestación más temprana y frecuente es la disfunción diastólica, aunque puede progresar a disfunción sistólica por infiltración miocárdica, dando lugar al síndrome de corazón rígido; además, compromete al sistema de conducción. Se debe sospechar en cualquier paciente con cardiomegalia no explicada por otra causa. Dentro de las herramientas diagnósticas se debe tener en cuenta la relación voltaje/ masa. La biopsia endomiocárdica es útil aunque no siempre positiva para la comprobación histológica. El tratamiento es de soporte y algunos casos se benefician del trasplante hepático.
Palabras clave: cardiomiopatía restrictiva, amiloidosis cardiaca, marcapasos.
Amyloidosis is an infiltrative systemic disease that may involve the heart. It has a genetic etiology and is an important cause of restrictive cardiomyopathy. It may involve all heart structures but has a great affinity for myocardial tissue. Diastolic dysfunction is the most early and frequent manifestation, although due to myocardial infiltration, it may progress to systolic dysfunction, resulting in a rigid heart syndrome. There is also an involvement of the conducting system. The condition may be suspected in any patient with cardiomegalia of unexplained cause. Among the diagnostic tools, the voltage/mass relation may be kept in mind. Endomyocardial biopsy is useful although it is not always positive through histological verification. The treatment consists of supportive measures and selected cases may benefit with hepatic transplantation.
Key words: restrictive cardiomyopathy, cardiac amyloidosis, pacemakers.
Casos clínicos
Se recibieron dos pacientes, uno de género masculino y otro de género femenino, con edades de 73 y 61 años, quienes consultaron por síntomas de astenia, adinamia y fatigabilidad y presentaron episodios de fibrilación auricular paroxística. En uno de ellos se documentó disfunción sinusal avanzada y en el otro bloqueo aurículo-ventricular intermitente. En ambos, el electrocardiograma demostró bajo voltaje generalizado.
En el ecocardiograma se evidenció hipertrofia ventricular izquierda con signos de refringencia que sugirieron enfermedad infiltrativa; en ambos la fracción de expulsión fue normal y se demostró disfunción diastólica. Se realizó tomografía axial computarizada (TAC) multidetector de tórax, la cual mostró áreas de menor densidad en el corazón que son compatibles con depósitos de amiloide (Figura 1). La TAC de abdomen no evidenció infiltración en vísceras extracardiacas. Uno de los pacientes tenía biopsias de grasa periumbilical y de recto, negativas para depósito de amiloide. Se descartaron causas secundarias de amiloidosis. A ambos se les implantaron marcapasos bicamerales; durante este procedimiento se demostró una onda P y R de muy bajo voltaje, con umbrales normales. Los dos pacientes desarrollaron incremento importante de los umbrales, por lo que se llevaron a reposición de electrodos auriculares. El diagnóstico de amiloidosis cardiaca se realizó por biopsia endomiocárdica en ambos pacientes.
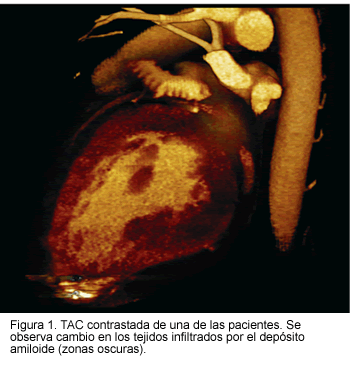
Uno de los pacientes murió un año después del diagnóstico de amiloidosis cardiaca por falla cardiaca, refractaria al tratamiento médico convencional. El otro paciente se llevó a transplante hepático con evolución satisfactoria y a la fecha se reporta buena evolución.
Introducción
El término amiloidosis hace referencia al depósito tisular extracelular de fibras compuestas de varias subunidades de bajo peso molecular (5 a 25 kD) de una variedad de proteínas séricas. El depósito amiloideo puede ocurrir en una variedad de órganos entre los que se incluyen corazón, riñón e hígado, que son responsables a menudo de la morbimortalidad de la enfermedad.
Las dos formas más comunes de amiloidosis son: amiloidosis primaria (AL), en la que la proteína amiloidea está compuesta por fragmentos monoclonales de cadenas livianas, lo cual es una discrasia de células plasmáticas; y amiloidosis secundaria (AA), que se caracteriza por fragmentos de proteína amiloidea A, la cual se asocia con desórdenes inflamatorios crónicos.
Entre las causas genéticas de amiloidosis, las mutaciones en el gen para la transtiretina (también llamada prealbúmina), son las que más se asocian con enfermedad cardiaca; con algunas mutaciones los depósitos amiloideos se limitan clínicamente al miocardio.
Importancia del tipo de amiloidosis
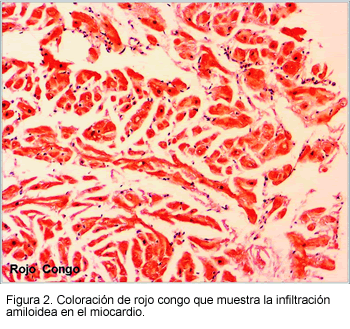
Patológicamente la cardiomiopatía amiloidea se define por la deposición amiloidea en el miocardio (Figura 2) y clínicamente por signos de disfunción miocárdica o del sistema de conducción. La evidencia clínica de compromiso cardiaco ocurre hasta en 50% de los pacientes con AL, en 10% con AA y en menos del 5% con síndromes familiares (1, 2).
Amiloidosis primaria comparada con otros tipos de amiloidosis
El compromiso cardiaco en amiloidosis primaria a menudo lleva a disfunción cardiaca significativa, mientras que la amiloidosis secundaria y las formas familiares, tiene un compromiso más leve (1-4). Esta distinción se ilustra en una revisión de 36 pacientes con cardiomiopatía amiloidea: 12 con forma familiar y 24 con amiloidosis secundaria (3). Aunque los dos grupos tienen similares hallazgos ecocardiográficos, las tasas de supervivencia a un año son de 92% y 38% respectivamente, y todas las muertes son de origen cardiaco (Figura 3).
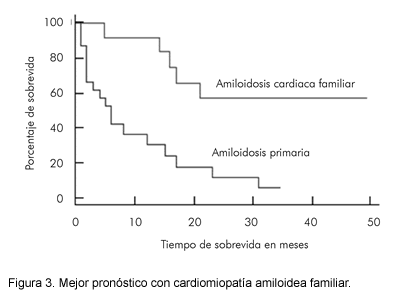
La diferencia en el pronóstico refleja el grado variable de la infiltración amiloidea del miocardio. Habitualmente, se observan depósitos difusos extensos en pacientes con AA, mientras que los focos aislados esparcidos son más característicos de otros tipos de amiloidosis. Otro posible factor que contribuye a la diferencia en el pronóstico, es que las características bioquímicas de los distintos tipos de fibrillas amiloideas tienen variados efectos patológicos en el miocardio (3).
Mutaciones de la transtiretina
Hay una variabilidad en la severidad relativa de la enfermedad cardiaca y extracardiaca entre los pacientes con diferentes mutaciones del gen encargado de codificar la transtiretina (TTR), en la que hay mayor compromiso en la población negra debido, en parte, a la presencia de un alelo de la TTR amiloidogénico heterocigoto, en el cual la isoleucina se sustituye por valina en la posición 122 (Ile 122). Este alelo no está presente en ningún paciente de raza blanca con amiloidosis cardiaca (5).
Reportes iniciales sugerían que la amiloidosis cardiaca senil, raras veces era una causa de morbimortalidad significativa (6, 7); sin embargo, ésta puede asociarse con fibrilación auricular, daño en el sistema de conducción y falla cardiaca (5).
Correlaciones clinicopatológicas
Los estudios con necropsia de amiloidosis cardiaca, muestran un miocardio engrosado con crecimiento ventricular y auricular. La inspección macroscópica revela una consistencia cauchosa y la presencia de trombos intracardiacos (8).
El examen microscópico del miocardio muestra depósitos hialinos que predominan en el espacio extracelular. Las fibrillas ligan la coloración de rojo congo (llevando a una birrefringencia verde bajo luz polarizada) (Figura 4).
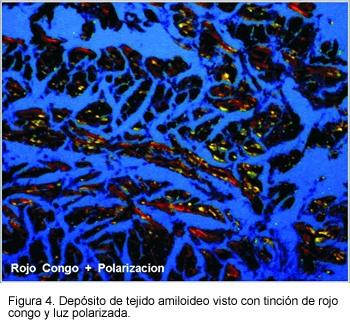
Desde el punto de vista clínico, el compromiso del miocardio ventricular y del sistema de conducción es lo más importante. No obstante, pueden encontrarse depósitos amiloideos clínicamente significativos en las siguientes localizaciones (9):
- Arterias coronarias intramiocárdicas y epicárdicas.
- Miocardio auricular.
- Válvulas cardiacas.
- Pericardio.
La acumulación de tejido amiloideo en una o más de estas áreas, puede llevar a distintas presentaciones clínicas.
Disfunción miocárdica
Los depósitos amiloideos difusos pueden llevar a daño en la función miocárdica por tres formas diferentes:
- Reemplazo del tejido miocárdico funcional con proteína amiloidea, lo cual lleva a reducción en la función sistólica (10).
- Infiltración difusa del miocardio ventricular izquierdo con fibrillas inertes y rígidas, que lleva a daño en la relajación y disfunción diastólica (10, 11). En los pacientes que aún preservan la función sistólica, esta anormalidad puede ser difícil de distinguir de la pericarditis constrictiva.
En algunos casos de marcada infiltración amiloidea, la combinación de un patrón de llenado restrictivo severo y una contracción ventricular izquierda reducida, pueden causar el llamado «síndrome de corazón rígido» (12). Este cuadro clínico es probablemente único para la amiloidosis.
Desde el punto de vista patológico, las características bioquímicas de las cadenas livianas pueden ser importantes. En un modelo experimental en el que se usaron corazones murinos aislados, la infusión de cadenas livianas de pacientes con AL que tenía compromiso cardiaco severo, deterioró de manera marcada la relajación ventricular sin afectar la función sistólica (11). Este hallazgo es similar al que se observa en pacientes con mieloma múltiple, en el cual las características bioquímicas de las cadenas livianas determinan el tipo de enfermedad renal y extrarrenal.
Anormalidades electrofisiológicas
En la enfermedad cardiaca amiloidea es común aceptar el sistema de conducción. Las anomalías no son a menudo clínicamente significativas, pero pueden ocurrir disturbios en la conducción y arritmias serias, incluso muerte súbita (5, 13, 14), así como fibrilación auricular, común en casos avanzados.
Aunque el nodo sinusal puede involucrarse en forma patológica (15), la función anormal parece ser más común en el sistema His-Purkinje. Esto se ilustró en un estudio electrofisiológico de 25 pacientes con AL y compromiso cardiaco (13). La función sinusal y nodal auriculoventricular se preservó en muchos pacientes, pero los tiempos de conducción infra hisianos estuvieron usualmente prolongados, de tal forma que 13 pacientes tuvieron intervalos HV mayores de 55 mseg y 12 pacientes mostraron intervalos HV mayores de 80 mseg. La prolongación del intervalo HV puede no sospecharse clínicamente si el QRS es estrecho en el electrocardiograma (EKG) de superficie (Figura 5).
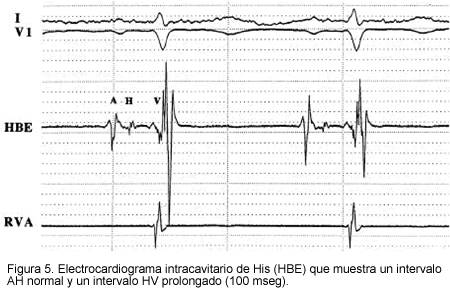
El compromiso del sistema Purkinje y del miocardio ventricular, puede llevar a activación miocárdica retardada, la cual puede detectarse en el EKG de actividad promediada (14). En un reporte de 133 pacientes con AL, los potenciales tardíos en el EKG de señal promediada fueron significativamente más frecuentes en pacientes con evidencia ecocardiográfica de compromiso amiloideo cardiaco (31% vs. 9% en aquellos con EKG normal). Tanto la prolongación del intervalo HV como los potenciales tardíos, se asocian independientemente con un alto riesgo de muerte súbita cardiaca (13, 14).
Manifestaciones clínicas
La presentación clínica de la enfermedad es usualmente dominada por falla cardiaca del lado derecho. Estas manifestaciones incluyen edema periférico, elevación de la presión venosa yugular, hipotensión ortostática (como resultado del inadecuado llenado ventricular y presencia de neuropatía autonómica). Hay también presencia de bajo gasto cardiaco con disfunción renal, daño en la perfusión orgánica, fatiga y edema pulmonar. El examen físico puede revelar soplo sistólico (debido a la regurgitación mitral y tricúspide) y baja presión de pulso (debido al bajo volumen latido).
El síncope es un síntoma común, multifactorial, dentro del que cabe destacar bradiarritmias y/o taquiarritmias e hipotensión postural (16).
El compromiso sintomático del sistema de conducción que requiere un marcapaso permanente es poco común (17).
Los pacientes con depósito amiloideo extenso en las arterias coronarias pueden desarrollar síntomas de angina típica (18-21). Los vasos involucrados son típicamente pequeños e intramiocárdicos, no epicárdicos; como resultado, la angiografía coronaria en estos pacientes es normal.
Raras veces, la acumulación amiloidea en el septo puede simular cardiomiopatía hipertrófica (22), mientras que los depósitos en el pericardio pueden producir taponamiento cardiaco (23, 24).
Diagnóstico
La presencia de amiloidosis debe descartarse en cualquier paciente con cardiomegalia no explicada, falla cardiaca, proteinuria severa o hepatomegalia. Hallazgos como anemia, dolor óseo o hipercalcemia sugieren AL con mieloma múltiple; mientras una historia de enfermedad inflamatoria crónica, como artritis reumatoidea, sugiere AA.
El diagnóstico de amiloidosis se confirma por la observación de depósitos amiloideos tisulares en panículo adiposo abdominal, recto o riñón. Los depósitos amiloideos en el corazón pueden llevar a anomalías detectables con varios exámenes.
Electrocardiografía
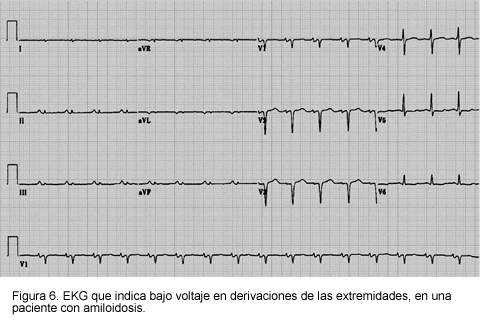
El hallazgo más común en amiloidosis cardiaca es el bajo voltaje en las derivaciones de las extremidades y precordiales; éste ocurre en aproximadamente 50% de los pacientes, particularmente en aquellos con AL (1, 3, 25). Otros cambios que pueden ocurrir incluyen anomalías de la conducción (bloqueo auriculoventricular de II y III grado), fibrilación auricular y patrones de seudoinfarto. Todas estas anomalías son no específicas.
Ecocardiografía
La disfunción diastólica es el hallazgo mas común, más temprano y más importante en amiloidosis cardiaca. Ésta es evidente antes de la disfunción sistólica en aquellos pacientes que llegan a tener falla de bomba. También se puede evidenciar disfunción diastólica ventricular derecha (26). En casos de enfermedad avanzada, la amiloidosis cardiaca es una causa de cardiomiopatía restrictiva que simula pericarditis constrictiva. La evaluación Doppler de las velocidades de flujo sanguíneo transmitral y transtricúspide, muestra un patrón de llenado restrictivo con fuerte dominancia de la onda E y corto tiempo de desaceleración.
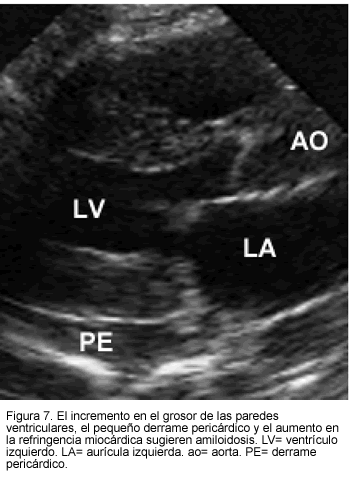
En pacientes con amiloidosis manifiesta, la ecocardiografía frecuentemente muestra engrosamiento simétrico de las paredes ventriculares izquierdas (27), aunque hay casos de compromiso septal asimétrico que produce una apariencia similar a la cardiomiopatía hipertrófica obstructiva (22). También se pueden observar cámaras ventriculares pequeñas, engrosamiento del septo auricular y dilatación auricular (28).
El hallazgo ecocardiográfico más distintivo en amiloidosis cardiaca, es la apariencia granular del miocardio, la cual es el resultado de la presencia de nódulos de colágeno y amiloide en el corazón (25, 27, 29). No obstante, cuando éste se halla solo, es relativamente no específico ya que otras enfermedades infiltrativas también lo pueden generar.
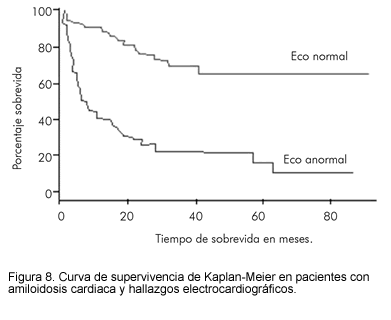
La ecocardiografía también provee información pronóstica, como se demostró en una serie hecha por Dubrey y colaboradores(14) en la cual en 133 pacientes con AL la presencia de muerte cardiaca fue más común en 77 pacientes con ecocardiograma anormal que mostraba evidencia de depósito amiloideo, en comparación con aquellos que tenían ecocardiograma normal (Figura 7)
.
Relación voltaje-masa
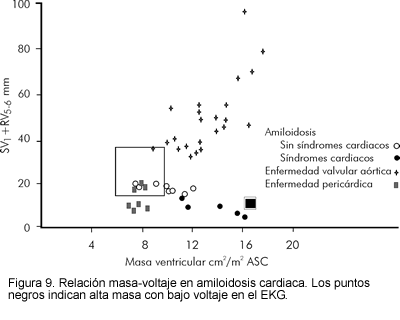
Las limitaciones de los exámenes antes mencionados para el diagnóstico de amiloidosis cardiaca, llevaron a la evaluación de otras modalidades, una de las cuales es la relación voltaje-masa cardiaca. La combinación de una masa ventricular incrementada (medida ecocardiográficamente) con reducción en el voltaje electrocardiográfico, es única para esta cardiomiopatía infiltrativa (25, 30, 31). Otras enfermedades que se caracterizan por bajo voltaje en el EKG (taponamiento, derrame pericárdico, enfisema) no se asocian con incremento en la masa cardiaca, mientras que enfermedades con masa cardiaca incrementada (hipertensión, hipertrofia) usualmente no se asocian con bajo voltaje en el EKG (Figura 9).
Biopsia endomiocárdica
Este examen permite observar depósitos amiloideos en los pacientes con amiloidosis sistémica documentada y sospecha de compromiso cardiaco. En dos estudios retrospectivos la biopsia miocárdica fue positiva en 40 de 41 pacientes, lo cual sugiere que este procedimiento es altamente sensible en pacientes seleccionados de manera adecuada (32, 33).
Las indicaciones para la realización de biopsia miocárdica por posible amiloidosis cardiaca, no están aún bien definidas; sin embargo, este procedimiento puede ser útil cuando hay un paciente con cardiomiopatía restrictiva la cual no puede ser diferenciada por otros métodos de pericarditis constrictiva. Además, se debe contemplar su realización cuando los hallazgos positivos alterarán la toma de decisiones en el manejo clínico.
Tratamiento
El tratamiento de la amiloidosis cardiaca sintomática es usualmente inefectivo y por lo general consiste en medidas de soporte.
Amiloidosis primaria
En estos pacientes se reporta un promedio en la supervivencia entre 6 y 9 meses cuando desarrollan falla cardiaca (34, 35) y de 1,1 años en aquellos con cualquier signo de compromiso cardiaco. La presencia de dilatación desproporcionada del ventrículo derecho puede asociarse con un pobre pronóstico (supervivencia en promedio de 4 meses) (28).
área ventricular fin diástole izquierda
área ventricular fin diástole derecha
Dilatación VD desproporcionada.
Varios reportes que usaron quimioterapia (melfalán-prednisona) mostraron resultados favorables en algunos pacientes (36-38).
Trasplante hepático o cardiaco
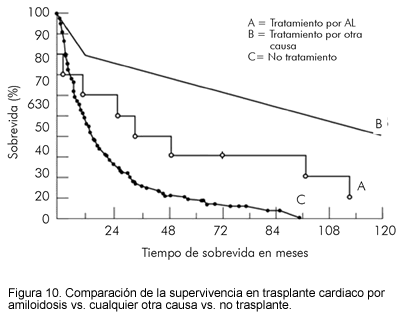
El transplante cardiaco no se ha utilizado como rutina en pacientes con AA o AL debido a que frecuentemente éstos tienen depósito amiloideo extracardiaco en forma severa, a que pueden progresar a mieloma múltiple y por la posibilidad de recurrencia en el corazón trasplantado. Pese a ello, hay reportes anecdóticos que sugieren beneficio temporal del trasplante cardiaco (39-41) (Figura 10).
El trasplante hepático puede ser curativo en casos de amiloidosis familiar debido a un defecto en la TTR.
Conclusión
El compromiso cardiaco de la amiloidosis debe sospecharse en todos los pacientes con cardiomiopatía restrictiva. Éstos desarrollan disfunción diastólica temprana y anomalías no específicas en el sistema de conducción.
Los hallazgos ecocardiográficos y electrocardiográficos son altamente sugestivos. El papel de la biopsia endomiocárdica aún no se define por completo. Las modalidades terapéuticas disponibles son sólo medidas de soporte.
Bibliografía
1. Gertz MA, Kyle RA. Primary systemic amyloidosis. A diagnostic primer. Mayo Clin Proc 1989; 64:1505. [ Links ]
2. Kyle RA. Amyloidosis. Circulation 1995; 91:1269. [ Links ]
3. Dubrey SW, Skinner M, LaValley M, et al. Familial and primary (AL) cardiac amyloidosis: echocardiographically similar diseases with distinctly 5 different clinical outcomes. Heart 1997; 78: 74. [ Links ]
4. Gertz A, Kyle RA, Thibodeau SN. Familial amyloidosis: A study of 52 North American-born patients examined during a 30 year period. Mayo Clin Proc 1992; 67:428. [ Links ]
5. Jacobson DR, Pastore RD, Yaghoubian R, et al. Variant sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N Engl J Med 1997; 336: 466. [ Links ]
6. Cornwell GG, Murdoch BS, Kyle RA, et al. Frequency and distribution of senile cardiovascular amyloid. Am J Med 1983; 75: 618. [ Links ]
7. Olson LJ, Gertz MA, Edwards WD, et al. Senile cardiac amyloidosis with myocardial dysfunction. Diagnosis by endomyocardial biopsy and immunohistochemistry. N Engl J Med 1987; 317: 738. [ Links ]
8. Roberts WC, Waller BF. Cardiac amyloidosis causing cardiac dysfunction: analysis of 54 necropsy patients. Am J Cardiol 1983; 52: 137. [ Links ]
9. Smith TJ, Kyle RA. Clinical significance of histological patterns of cardiac amyloidosis. Mayo Clin Proc 1984; 59: 547. [ Links ]
10. Swanton RH, Brooksby IAB, Davies MJ, et al. Systolic and diastolic ventricular function in cardiac amyloidosis. Am J Cardiol 1977; 39: 658. [ Links ]
11. Liao R, Jain M, Teller P, et al. Infusion of light chains from patients with cardiac amyloidosis causes diastolic dysfunction in isolated mouse hearts. Circulation 2001; 104: 1594. [ Links ]
12. Chew C, Ziady GM, Raphael MJ, Oakley CM. The functional defect in amyloid heart disease. The «stiff heart» syndrome. Am J Cardiol 1975; 36:43 (abstract). [ Links ]
13. Reisinger J, Dubrey SW, Lavalley M, et al. Electrophysiologic abnormalities in AL (primary) amyloidosis with cardiac involvement. J Am Coll Cardiol 1997; 30:1046. [ Links ]
14. Dubrey SW, Bilazarian S, LaValley M, et al. Signal averaged electrocardiography in patients with AL (primary) amyloidosis. Am Heart J 1997; 134: 994. [ Links ]
15. Ridolfi RL, Bulkley BH, Hutchins GM. The conduction system in cardiac amyloidosis. Am J Med 1977; 62: 677. [ Links ]
16. Chamarthi B, Dubrey SW, Cha K, et al. Features and prognosis of exertional syncope in light-chain associated AL cardiac amyloidosis. Am J Cardiol 1997; 80: 1242. [ Links ]
17. Mathew V, Olson LJ, Gertz MA, et al. Symptomatic conduction system disease in cardiac amyloidosis. Am J Cardiol 1997; 80: 1491. [ Links ]
18. Al Suwaidi J, Velianou JL, Gertz MA, et al. Systemic amyloidosis presenting as angina pectoris. Ann Intern Med 1999; 131: 838. [ Links ]
19. Mueller PS, Edwards WD, Gertz MA. Symptomatic ischemic heart disease resulting from obstructive intramural coronary amyloidosis. Am J Med 2000; 109: 181. [ Links ]
20. Smith RR, Hutchins GM. Ischemic heart disease secondary to amyloidosis of intramyocardial arteries. Am J Cardiol 1979; 44: 413. [ Links ]
21. Hongo, M, Yamamoto H, Kohda T, et al. Comparison of electrocardiographic findings in patients with AL (primary) amyloidosis and in familial amyloid polyneuropathy and anginal pain and their relation to histopathologic findings. Am J Cardiol 2000; 85: 849. [ Links ]
22. Sedlis SP, Saffitz JE, Schwob VS, Jaffe AS. Cardiac amyloidosis simulating hypertrophic cardiomyopathy. Am J Cardiol 1984; 53: 969. [ Links ]
23. Brodarick S, Paine R, Higa E, Carmichael KA. Pericardial tamponade, a new complication of amyloid heart disease. Am J Med 1982; 73: 133. [ Links ]
24. Daubert JP, Gaede J, Cohen HJ. A fatal case of constrictive pericarditis due to marked, selective pericardial accumulation of amyloid. Am J Med 1993; 94: 335. [ Links ]
25. Rahman JE, Helou EF, Gelzer-Bell R, et al. Noninvasive diagnosis of biopsy-proven cardiac amyloidosis. J Am Coll Cardiol 2004; 43: 410. [ Links ]
26. Klein AL, Hatle LK, Burstow DJ, et al. Comprehensive Doppler assessment of right ventricular diastolic function in cardiac amyloidosis. J Am Coll Cardiol 1990; 15: 99. [ Links ]
27. Siqueira-Filho AG, Cunha CLP, Tajik AJ, et al. M-mode and two-dimensional echocardiographic features in cardiac amyloidosis. Circulation 1981; 63: 188 (abstract). [ Links ]
28. Patel AR, Dubrey SW, Mendes LA, et al. Right ventricular dilatation in primary amyloidosis: an independent predictor of survival. Am J Cardiol 1997; 80: 486. [ Links ]
29. Falk RH, Plenn JF, Deering T, et al. Sensitivity and specificity of echocardiographic features of cardiac amyloidosis. Am J Cardiol 1987; 59: 418 (abstract). [ Links ]
30. Simons M, Isner JM. Assessment of relative sensitivities of non-invasive tests for cardiac amyloidosis in documented cardiac amyloidosis. Am J Cardiol 1992; 69: 425 (abstract). [ Links ]
31. Carroll JD, Gaasch WH, McAdam KPWJ. Amyloid cardiomyopathy: characterization by a distinctive voltage/mass relation. Am J Cardiol 1982; 49: 9. [ Links ]
32. Pellikka PA, Holmes DR Jr., Edwards WD, et al. Endomyocardial biopsy in 30 patients with primary amyloidosis and suspected cardiac involvement. Arch Intern Med 1988; 148:662 (abstract). [ Links ]
33. Arbustini E, Merlini G, Gavazzi A, et al. Cardiac immunocyte-derived amyloidosis: an endomyocardial biopsy study in 11 patients. Am Heart J 1995; 130: 528. [ Links ]
34. Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol 1995; 32: 45. [ Links ]
35. Dubrey SW, Cha K, Anderson J, et al. The clinical features of immunoglobulin light-chain (AL) amyloidosis with heart involvement. QJM 1998; 91: 141. [ Links ]
36. Kyle RA, Gertz MA, Greipp PR, et al. A trial of three regimens for primary amyloidosis: colchicine alone, melphalan and prednisone, and melphalan, prednisone, and colchicine. N Engl J Med 1997; 336: 1202. [ Links ]
37. Grogan M, Gertz MA, Kyle RA, Tajik AJ. Five or more years of survival in patients with primary systemic amyloidosis and biopsy-proven cardiac involvement. Am J Cardiol 2000; 85: 664. [ Links ]
38. Dubrey S, Mendes L, Skinner M, Falk RH. Resolution of heart failure in patients with AL amyloidosis. Ann Intern Med 1996; 125: 481. [ Links ]
39. Pelosi F, Capehart J, Roberts WC. Effectiveness of cardiac transplantation for primary (AL) cardiac amyloidosis. Am J Cardiol 1997; 79: 532. [ Links ]
40. Dubrey SW, Burke MM, Khaghani A, et al. Long term results of heart transplantation in patients with amyloid heart disease. Heart 2001; 85: 202. [ Links ]
41. Stangou AJ, Hawkins PN, Heaton ND, et al. Progressive cardiac amyloidosis following liver transplantation for familial amyloid polyneuropathy. Transplantation 1998; 66: 229. [ Links ]

