










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Cardiología
Print version ISSN 0120-5633
Rev. Col. Cardiol. vol.14 no.1 Bogota Jan./Feb. 2007
Clínica de Marly, Bogotá, DC., Colombia.
Correspondencia: Vanessa Torres Gómez, MD. Clínica de Marly, Calle 50 No. 9- 67 Oficina 605, Teléfono: 343 6600. Extensión: 1606 ó 2611. Correo electrónico: vanetorres16@yahoo.com.
Recibido: 15/09/06. Aprobado: 04/02/07.
La hipertensión arterial pulmonar es una entidad recientemente clasificada como idiopática y secundaria, de acuerdo con su etiología. Se caracteriza por el aumento de la presión de la arteria pulmonar que progresivamente ocasiona dilatación, falla ventricular derecha y muerte prematura. La incidencia de la enfermedad es baja, pero tiene implicaciones fisiopatológicas muy severas. Se han determinado causas endógenas y exógenas que contribuyen al desarrollo de la enfermedad. El tratamiento se enfoca en mejorar la función ventricular para lo cual se emplean vasodilatadores y nuevos fármacos dirigidos a inducir la relajación del músculo liso vascular. Además, se utiliza oxígeno si mejora el estado del paciente, y anticoagulación oral.
Palabras clave: hipertensión pulmonar idiopática, resistencia vascular pulmonar, falla ven-tricular derecha.
Pulmonary arterial hypertension is a recently classified as primary and secondary entity according to its etiology. It is characterized by increased pulmonary artery pressure that results in progressive dilation, right ventricular failure and premature death. The incidence of this disease is low, but has severe physiopathologic implications. Many endogenous and exogenous causes that contribute to the development of the disease have been determined. Treatment is focused on improving ventricular function for which purpose vasodilators and new drugs directed to induce vascular smooth muscle relaxation are used. Besides, oxygen is used when it improves patients condition, as well as oral anticoagulation.
Key words: idiopatic pulmonary hypertension, pulmonary vascular resistance(PVR), right ventricular failure.
Definición
La hipertensión pulmonar se define como un incremento progresivo de las resistencias vasculares pulmonares, que conlleva un aumento de la presión en la arteria pulmonar que genera falla ventricular derecha y como consecuencia muerte prematura (1).
Se caracteriza por el aumento de la presión media de la arteria pulmonar mayor o igual a 25 mm Hg en reposo, o de 30 mm Hg durante el ejercicio (2-4). La hipertensión pulmonar idiopática es una condición que se caracteriza por el aumento significativo de la presión de la arteria pulmonar, sin una causa demostrable.
La hipertensión pulmonar se clasifica como idiopática (primaria en clasificaciones anteriores) y asociada con otras condiciones o secundaria, dependiendo de la presencia o ausencia de factores de riesgo o causas identificables etiológicas como enfermedades del tejido conectivo, cardiopatías congénitas, hipertensión portal e infección por virus de inmunodeficiencia adquirida; todas éstas con alteraciones similares en la microvasculatura pulmonar, de tipo obstructivo (5-7).
El diagnóstico de hipertensión pulmonar primaria es de exclusión, cuando se han estudiado otras entidades sin encontrar la causa de la misma. En 1998 durante la Segunda Reunión Mundial de Hipertensión Pulmonar en Evian (Francia), se propuso trabajar en una clasificación de hipertensión pulmonar basada en hallazgos clínicos, lo que la haría más práctica para su interpretación. Fue así como el objetivo de la "Clasificación Evian" se dirigió a agrupar las enfermedades por categorías de acuerdo con semejanzas en cuanto a su mecanismo fisiopatológico, presentaciones clínicas y opciones terapéuticas.
En 2003 durante el Tercer Consenso Mundial de Hipertensión Pulmonar en Venecia (Italia) se decidió realizar algunas modificaciones, dentro de las que se eliminó el término "Hipertensión pulmonar primaria" y se reemplazó por «Hipertensión pulmonar idiopática y se reclasificaron las enfermedades veno-oclusivas, congénitas y aquellas derivadas de malformaciones vasculares pulmonares (8).
Incidencia
De acuerdo con estudios norteamericanos, se considera que la incidencia de hipertensión pulmonar primaria es muy baja, uno a dos casos por cada millón de habitantes de la población general (9). Se ha observado que el uso de supresores del apetito incrementa el riesgo de hipertensión pulmonar primaria y más aún, si se han usado de forma continua por más de tres meses. Los medicamentos asociados son la fenfluramina y la dexfenfluramina, ambos inhibidores de la recaptación de serotonina, y afectan de manera independiente el índice de masa corporal, lo que indica que la obesidad per se, no aumenta el riesgo de desarrollar la enfermedad. Debido al poco porcentaje de presentación, se ha propuesto la necesidad de una predisposición genética o un rasgo heredado de forma mendeliana, caso presentado en la hipertensión pulmonar familiar que tiene una herencia autonómica dominante, al parecer por alteraciones del cromosoma X (10).
Fisiopatología
Se han identificado características endógenas y exógenas importantes, que generan aumento de las resistencias vasculares de la arteria pulmonar.
Características endógenas
La primera y tal vez predominante, tiene que ver con un disbalance entre sustancias vasodilatadoras-vasoconstrictoras, en las que sobresale este último mecanismo; además hay proliferación de músculo liso y del endotelio y posterior trombosis microvascular.
Esta alteración de la homeostasis de sustancias vasculo-efectoras es la responsable de la lesión y disfunción endotelial. Es así como se ha evidenciado aumento de tromboxano A2, potente vasoconstrictor y facilitador de la agregación plaquetaria, y disminución importante en los niveles de prostaciclina con su efecto vasodilatador antagónico. En la orina de pacientes con hipertensión pulmonar los niveles de metabolitos de prostaciclina se encuentran disminuidos (6 keto-prostaciclin-F 2a) mientras que los niveles de metabolitos de tromboxano A2 (tromboxano B2) se encuentran elevados (11).
Los niveles elevados de endotelina en pacientes con hipertensión pulmonar, producen vasoconstricción y adicionalmente estimulan la proliferación de músculo liso. La serotonina produce efectos vasoconstrictores similares en contraste con los del óxido nítrico, un potente vasodilatador e inhibidor de la activación plaquetaria, pero en pacientes con hipertensión pulmonar idiopática los niveles son bajos (12).
En conejos el péptido intestinal vasoactivo se ha identificado como potente vasodilatador que genera disminución de las resistencias de la arteria pulmonar, y actualmente está en estudio en pacientes con hipertensión pulmonar en quienes se ha encontrado disminución de esta sustancia en suero, por lo que en estos pacientes se ha planteado el tratamiento inhalado de péptido intestinal vasoactivo (13).
Características exógenas
En forma general, la hipoxia induce vasodilatación sistémica; sin embargo, en los vasos pulmonares induce vasoconstricción. Esto ocurre debido a que la hipoxemia genera alteración del voltaje de la membrana celular y de los canales de potasio con despolarización de la membrana y aumento intracelular de los niveles de calcio, lo cual produce vasoconstricción, remodelación y proliferación de células de músculo liso, constituyendo un círculo permanente. Aún así, la hipoxia como tal, no es el desencadenante de la hipertensión, sino que contribuye a perpetuar la entidad una vez se ha instaurado.
Los anorexígenos con compuestos de fenfluramina o dexfenfluramina, se relacionan con aumento del riesgo para desarrollar hipertensión arterial pulmonar asociada con la dosis y duración. Usualmente, las primeras manifestaciones se aprecian hacia la tercera o cuarta semana de administración. Así mismo, el uso de cocaína y estimulantes del sistema nervioso central como las metanfetaminas, aumentan el riesgo de padecerla.
Manifestaciones clínicas
En pacientes con disnea, dolor torácico, síncope, debilidad, sensación de ahogo con el ejercicio, en quienes se haya descartado enfermedad cardiaca y pulmonar, sin otros signos específicos se debe sospechar hipertensión pulmonar y enfocar sus estudios a corroborar la enfermedad (14, 15). La angina en estos pacientes es el resultado de hipoperfusión subendocárdica secundaria al aumento de la tensión de la pared del ventrículo derecho y de la demanda de oxígeno. Sin embargo, se ha observado que un tronco pulmonar dilatado mayor de 40 mm de diámetro, genera compresión dinámica de la arteria coronaria izquierda y produce dolor torácico (16, 17).
En el examen físico es característico el aumento de la intensidad del segundo ruido en el foco pulmonar y en casos severos se puede palpar la arteria pulmonar en el segundo espacio intercostal izquierdo, donde es factible percutir un tono mate dado por la arteria pulmonar dilatada. Es frecuente encontrar soplo sistólico de regurgitación tricúspide, ingurgitación yugular con onda "v" prominentes debido a la insuficiencia tricúspide, y puede existir reflujo hepatoyugular. En casos más severos ingurgitación yugular bilateral, hepatomegalia, edema de miembros inferiores o ascitis (18).
Diagnóstico
En el estudio de la hipertensión pulmonar el objetivo principal es confirmar el diagnóstico. Para ello se realizan: electrocardiograma, radiografía de tórax, ecocardiograma, pruebas de función pulmonar, cateterismo cardiaco, estudios inmunológicos, entre otros (3).
Electrocardiograma
Es útil para evidenciar la severidad hemodinámica de la enfermedad. Se puede encontrar hipertrofia del ventrículo derecho, eje desviado a la derecha y dilatación auricular derecha (Figura 1). La hipertrofia del ventrículo derecho y la desviación del eje a la derecha, se pueden evidenciar en cerca del 80% de los pacientes con hipertensión pulmonar; sin embargo, la sensibilidad y especificidad del estudio sólo permiten un tamizaje de los cambios esperados en este tipo de patología. Un electrocardiograma normal nunca descarta la presencia de la enfermedad (19-21).
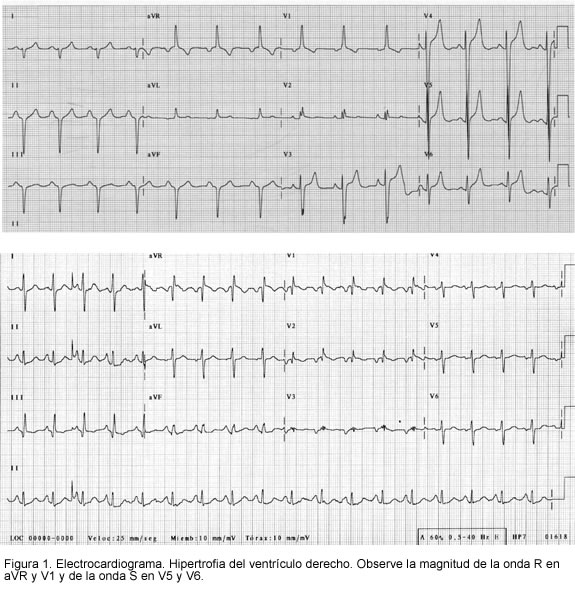
Radiografía de tórax
Se puede observar dilatación de la arteria pulmonar, en contraste con pérdida de los vasos sanguíneos periféricos y dilatación aurículo-ventricular derecha en casos severos de hipertensión pulmonar (Figura 2). En pacientes con hipertensión post-capilar es importante descartar enfermedad cardiaca. Una radiografía normal, no excluye la presencia de hipertensión pulmonar (3).
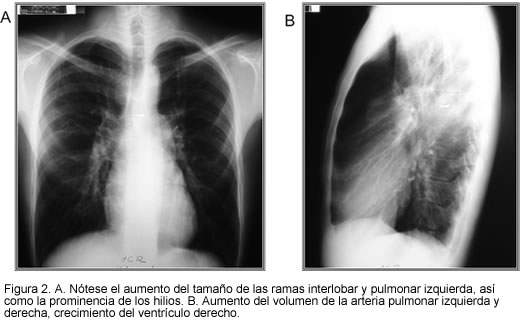
Ecocardiograma
El ecocardiograma es útil para demostrar hipertensión pulmonar y descartar patología estructural que cause la enfermedad. Para el diagnóstico de la hipertensión pulmonar, se deben tener en cuenta signos indirectos como dilatación e hipertrofia de las cavidades derechas, dilatación de la arteria pulmonar y aplanamiento del tabique interventricular que con frecuencia cambia la dirección de sus movimientos en sentido paradójico (Figura 3). Aunque se han descrito diversos métodos para cuantificar la presión pulmonar, el más práctico es medir el gradiente sistólico de regurgitación tricúspide a través de la ecuación de Bernaulli modificada (DP= 4xV2) y sumando la presión estimada para la aurícula derecha (41). De igual manera, se puede estimar la presión diastólica, si hay insuficiencia pulmonar, determinando el gradiente diastólico final.
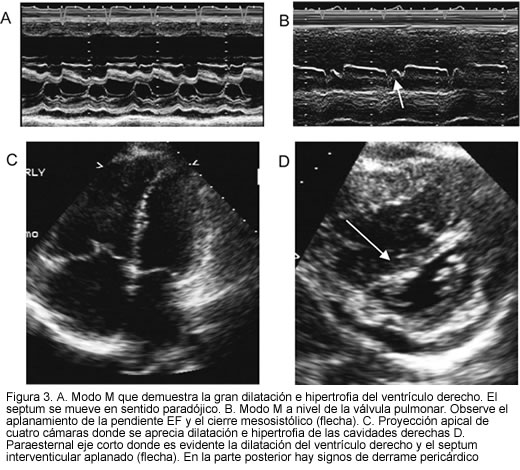
Para descartar patología estructural como comunicación interauricular o interventricular, ductus arterioso, estenosis mitral, etc. es importante hacer un estudio completo y detallado de todas las estructuras del corazón. En caso de duda se debe complementar con contraste salino, que puede poner de manifiesto cortocircuitos que no son evidentes en el ultrasonido, por ejemplo fístulas arteriovenosas extrapulmonares o comunicación interauricular alta del tipo de seno venoso (22-25).
Pruebas de función pulmonar
Estos estudios son importantes para descartar patología parenquimatosa pulmonar. En pacientes con enfermedad pulmonar obstructiva crónica, enfermedad intersticial pulmonar o fibrosis pulmonar y en las enfermedades intersticiales, puede presentarse hipertensión pulmonar, aunque ésta es menos severa que la que aparece en pacientes de etiología primaria.
En estos pacientes, las oximetrías nocturnas, pueden mostrar desaturaciones que corresponden a apnea obstructiva del sueño, que progresivamente agravan la hipertensión pulmonar debido a que la hipoxemia se comporta como un potente vasoconstrictor, empeorando la hipertensión pulmonar. Sin embargo en cerca del 75% de los pacientes con hipertensión pulmonar, ocurre hipoxemia nocturna sin apnea del sueño (26).
De igual manera, los estudios de ventilación/perfusión, las escanografías de tórax, la resonancia magnética nuclear entre otros, pueden contribuir a establecer causas de la enfermedad.
Tratamiento
El tratamiento de la hipertensión pulmonar se enfoca directamente en las causas de la enfermedad. Las estrategias terapéuticas se dirigen a mejorar la restricción en la circulación pulmonar e incrementar la demanda de oxígeno que empeora progresivamente la hipertensión pulmonar y la falla cardiaca derecha. Existen tres blancos a intervenir, la vía de la endotelina, el óxido nítrico y la prostaciclina, la anticoagulación y medidas de soporte general como la oxígenoterapia (27).
Manejo médico
Oxígeno: la hipoxemia en los pacientes con hipertensión pulmonar usualmente es entre leve y moderada. En pacientes con malformaciones congénitas, el cortocircuito derecha a izquierda es responsable de la hipoxemia, y ésta es directamente proporcional al tamaño del defecto. En la actualidad no existen estudios que apoyen o se opongan al uso de oxígeno dentro del manejo de estos pacientes, pero se ha documentado la necesidad de mantener cifras de saturación de oxígeno por encima de 90% (27).
Anticoagulantes: en los pacientes con hipertensión pulmonar es importante el manejo con anticoagulantes, debido al riesgo de eventos trombóticos en la microvasculatura, secundarios al desequilibrio entre tromboxanos y prostaciclinas, dilatación de cavidades derechas, estasis venosa y vida sedentaria entre otros. Varios estudios han demostrado hallazgos de eventos trombóticos en cadáveres de pacientes con hipertensión pulmonar. De igual modo, se ha determinado que el uso de warfarina disminuye el número de eventos trombóticos o microtrombóticos, disminuyendo así el riesgo de muerte. La dosis inicial es de 5 a 10 mg/día vía oral, pero el tratamiento se debe individualizar de acuerdo con la morbilidad asociada, con el objetivo de lograr mantener un INR de 2 a 2,5 como lo indica la mayor parte de la literatura (34, 35).
Calcioantagonistas: el uso de calcioantagonistas va dirigido a sus tradicionales efectos vasodilatadores. Se ha determinado aumento en la sobrevida de pacientes que se manejan con calcioantagonistas, con hallazgos de disminución de hipertrofia ventricular derecha y modulación de respuesta vasorreactiva en algunos pacientes susceptibles. Usualmente son necesarios los tratamientos prolongados y a dosis elevadas con calcioantagonistas para ver efectos favorables dentro del tratamiento. Los medicamentos de elección son diltiazem y nifedipino, y la escogencia de uno de ellos depende de la frecuencia cardiaca basal del paciente. Los pacientes con tendencia a presentar bradicardia, se favorecen de nifedipino y aquellos con taquicardia se benefician de diltiazem. El verapamilo no se usa por su importante efecto inotrópico negativo. Las dosis que han demostrado eficacia son elevadas; nifedipino de 120 a 240 mg/día, y diltiazem de 240 a 720 mg/día. Las situaciones que limitan el uso de dosis altas de calcioantagonistas tienen que ver con sus efectos hipotensores y con edemas periféricos (36, 37).
Bosentán: la endotelina estimula la proliferación de músculo liso vascular, induce fibrosis y produce vasoconstricción de las arteriolas pulmonares. El uso del Bosentán que es un antagonista selectivo de los receptores de endotelina modula esta serie de alteraciones fisiopatológicas. Se ha demostrado que a dosis de 125 mg dos veces al día mejora la hipertensión pulmonar. Es importante evaluar la función hepática (28)
Sildenafil: el óxido nítrico es reconocido como un vasodilatador potente con acción directa ya que relaja el músculo liso vascular, pero su duración de acción es fugaz. Se ha demostrado que su administración por vía inhalada disminuye en forma significativa la resistencia vascular pulmonar (42, 43). El sildenafil (inhibidor de la 5 fosfodiesterasa) incrementa la actividad endógena del óxido nítrico y produce un efecto vasodilatador directo. Actualmente se desarrollan estudios para corroborar su efectividad, efectos adversos y seguridad (29, 30).
Epoprostenol inhalado: la vía de la prostaciclina I², producto del ácido araquidónico, induce relajación del músculo liso vascular estimulando la vía del AMP cíclico, y adicionalmente es un potente inhibidor de la agregación plaquetaria. Estudios realizados con cohortes de pacientes revelan que a dosis de 100 a 150 mcg al día inhalado, mejora de manera significativa la capacidad pulmonar frente al ejercicio y el estado hemodinámico de los pacientes tratados, disminuyendo las resistencias vasculares pulmonares (31-33).
Manejo quirúrgico
Septostomía atrial: ésta crea un cortocircuito que descomprime el volumen y las presiones de las cavidades derechas. Es un manejo paliativo o en algunos casos en paso previo a la estabilización del paciente que va a ser llevado a trasplante de pulmón. En países en los que no hay disponibilidad de terapias médicas avanzadas, probablemente esta medida se convierta en una opción de tratamiento (38).
Trasplante de pulmón: está reservado para pacientes con franco deterioro de su estado general, en quienes no ha sido posible mejorar su condición con manejo médico. La sobrevida de pacientes con trasplante de pulmón usualmente es de 66% a 75% en el primer año. La mayoría de los centros de referencia hacen trasplante pulmonar bilateral en pacientes con hipertensión pulmonar (39, 40).
Conclusiones
La hipertensión pulmonar es una enfermedad grave, progresiva, que tiende a evolucionar hacia falla cardiaca y muerte prematura. Dado que los síntomas pueden ser ambiguos, es importante la sospecha clínica basada en factores de riesgo y hallazgos clínicos o paraclínicos. Se debe confirmar con electrocardiograma, radiografía de tórax, ecocardiograma y cateterismo cardiaco.
El tratamiento se basa en tratar la causa subyacente si ésta existe, oxígeno, anticoagulación y anticálcicos. Recientemente se han desarrollado nuevas opciones terapéuticas como óxido nítrico, sildenafil, bosentán y cirugía.
Bibliografía
1. Simonneau G, Galie N, Rubin L et al. Clinical classification of pulmonary arterial hypertension. J Am Coll Cardiol 2004; 43: S5-12. [ Links ]
2. Gaine SP, Rubin LJ. Primary pulmonary hypertension. Lancet 1999; 353: 74. [ Links ]
3. Lewis J, Rubin, David B, Badesch. Evaluation and management of the patient with pulmonary arterial hypertension. Ann Intern Med. 2005; 143: 282-292. [ Links ]
4. Harrison W, Farber L, et al, Pulmonary arterial hypertension. Mechanisms of disease. N Engl J Med 2004; 351: 1655-1665 [ Links ]
5. Galie N, Manes A, Uguccioni L et al. Primary pulmonary hypertension: insight into pathogenesis from epidemiology. Chest 1998; 114: 1845-94S. [ Links ]
6. Pietra GG, Edwards WD, Kay JM et al. Histopathology of primary pulmonary hypertension. A quantitative study of pulmonary blood vessels from 58 patients in the national heart lungs, and blood institute. Circulation 1989; 80: 1198-1206. [ Links ]
7. Pietra GG, Capron F, Stewart S, et al. Pathologic assessment of vasculopathies in pulmonary hypertension. J Am Coll of Cardiol. 2004: S25-32. [ Links ]
8. Fischman AP. Primary pulmonary arterial hypertension: a look back. J Am Coll Cardiol 204: 43: S2-4. [ Links ]
9. Abenhaim L, Moride Y, Brenot F, et al. Appetite suppresant drugs and the risk of primary pulmonary hypertension. N Engl J Med 1996; 335: 609-16. [ Links ]
10. Lewis J, Rubin MD. Primary pulmonary hypertension. Review article. N Engl J Med 1997: 336: 111-7. [ Links ]
11. Tuder RM, Cool CD, Geraci MW, et al. Prostacyclin synthase expression in decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med 1999; 159: 1925-32. [ Links ]
12. Mason NA. High expression of endothelial nitric oxide synthase in plexiforms lesions of pulmonary hypertension. J Pathol 1998; 185: 13-8. [ Links ]
13. Petkov V, Mosgoeller W, et al. Vasoactive intestinal peptide as a new rug or treatment of primary pulmonary hypertension. J Clin Invest 2003; 111: 1139-46. [ Links ]
14. Runo JR, Loyd, JE. Primary pulmonary hypertension. Lancet 2003; 361 (9368): 1533-44. [ Links ]
15. Peacock AJ. Primary pulmonary hypertension. Thorax 1999; 54 (12): 1107-18. [ Links ]
16. Mesquita SM, Castro CR, Ikari NM, Oliveira SA, Lopes AA. Likelihood of left main coronary artery compression based on pulmonary trunk diameter in patients with pulmonary hypertension. Am J Med 2004; 116 (6): 369-74. [ Links ]
17. Kawut SM, Silvestry FE, Ferrari VA, DeNofrio D, Axel L, et al. Extrinsic compression of the left main coronary artery by the pulmonary artery in patients with long-standing pulmonary hypertension. Am J Cardiol 1999; 83 (6): 984-6, A10. [ Links ]
18. Rich S. Primary pulmonary Hypertension. En: Harrison. Principles of Internal Medicine. 16th Edition. Disorders of respiratory system. Section 2. Diseases of respiratory System. Chapter 220. Primary Pulmonary Hypertension. Editorial McGraw-Hill, New York, 2001. p. 1403-1408. [ Links ]
19. Ahearn GS, Tapson VF, et al. Electrocardiography to define critical status primary pulmonary hypertension and pulmonary arterial hypertension secondary to collagen vascular disease. Chest 2002; 122: 524-7. [ Links ]
20. Bossone E, Paciocco G, Iarussi D, Agretto A, Iacono A, Gillespie BW et al. The prognostic role of the ECG in primary pulmonary hypertension. Chest 2002; 121 (2): 513-8. [ Links ]
21. Ahearn GS, Tapson VF, Rebeiz A, Greenfield JC Jr. Electrocardiography to define clinical status in primary pulmonary hypertension and pulmonary arterial hypertension secondary to collagen vascular disease. Chest 2002; 122 (2): 524-7. [ Links ]
22. Bosone E, Rubenfire M, Bach DS, Ricciardi M, Armstrong WF. Range of tricuspid regurgitation velocity at rest and during exercise in normal adult men: implications for the diagnosis of pulmonary hypertension. J Am Coll Cardiol 1999; 33: 1662-6. [ Links ]
23. Bossone E, Bodini BD, Mazza A, Allegra L. Pulmonary arterial hypertension: the key role of echocardiography. Chest 2005; 127 (5): 1836-43. [ Links ]
24. Hinderliter AL, Willis PW, Long W, Clarke WR, Ralph D, Caldwell EJ, et al. Frequency and prognostic significance of pericardial effusion in primary pulmonary hypertension. PPH Study Group. Primary pulmonary hypertension. Am J Cardiol 1999; 84 (4): 481-4, A10. [ Links ]
25. Ommen SR, Nishimura DG et al. Assessment of right atrial pressure with 2-dimensional and Doppler echocardiography: a simultaneous catheterizacion and echocardiographic study. Mayo Clin Proc 2000; 75: 24-9. [ Links ]
26. Rafanan AL, Golish JA, Dinner DS et al. Nocturnal hypoxemia is common in primary pulmonary hypertension. Chest 2001; 120: 894-9. [ Links ]
27. Marc Humbert, Oliver Sitbon, Gerald Simonneau. Treatment of pulmonary arterial hypertension. N Engl J Med 2004; 351: 1425-36. [ Links ]
28. Rubin LJ, Badesh DB, Barst RJ. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 2002; 346: 896-903. [ Links ]
29. Methta S. Sildenafil for pulmonary arterial hypertension: exciting, but protection required. Chest 2003; 123: 989-92. [ Links ]
30. Ghofrani HA, Wiedemann R, et al. Sildenafil for treatment of lung fibrosis and pulmonary hypertension: a randomised controlled trial. Lancet 2002; 360: 895-900. [ Links ]
31. Olschewski H, Simonneau G, Nazareno G, et al. Inhaled Iloprost for severe pulmonary hypertension. N Engl J Med 2002; 347: 322-9. [ Links ]
32. Mclaughlin V, Genther D, Panella M, et al. Reduction in pulmonary vascular resistance with long-term Epoprostenol (prostacyclin) therapy in primary pulmonary hypertension. N Engl J Med 1998: 338: 273-7. [ Links ]
33. Hoeper M, Schwarze M, Ehlerding S. et al. Long term treatment of primary pulmonary hypertension with aerosolized Iloprost, a prostacyclin analogue. N Eng J Med 2002; 342: 1886-70. [ Links ]
34. Fuster V, Steele PM, Edwards WD, Gersh BJ et al. Primary pulmonary hypertension: natural history and the importance of thrombosis. Circulation 1984; 70 (4): 580-7. [ Links ]
35. Kawut SM, Horn EM, Berekashvili KK, Garofano RP, Goldsmith RL, et al. New predictors of outcome in idiopathic pulmonary arterial hypertension. Am J Cardiol 2005; 95 (2): 199-203. [ Links ]
36. Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med 1992; 327 (2): 76-81. [ Links ]
37. Rich S, Brundage BH. High-dose calcium channel blocking therapy for primary pulmonary hypertension: evidence for log term reduction in pulmonary arterial pressure and regression of right ventricular hypertrophy. Circulation 1987; 76: 135-41. [ Links ]
38. Rothman A, Sklansky MS, Lucas VW, Kazan IA et al. Atrial septostomy as a bridge to lung trasplantation in patients with severe pulmonary hypertension. Am J Cardiol 1999; 84: 682-6. [ Links ]
39. Mendeloff EN, Meyers BF, Sundt TM, et al. Lung transplantation for pulmonary vascular disease. Ann Thorac Surg 2002; 73: 209-1799. [ Links ]
40. Pielsticker EJ, Martinez FJ, Rubenfire M. Lung and heart-lung transplant practice patterns in pulmonary hypertension centers. J Heart Lung Transplant 2001; 20: 1297-304. [ Links ]
41. Hatle L, Angelsen BAJ, Tromsdaal A. Noninvasive estimation of pulmonary artery systolic pressure with Doppler ultrasound. Br Heart J 1981; 45: 157-67. [ Links ]
42. Inglessis I, Shin JT, Lepore JJ, Palacios IF, Zapol WM, Bloch KG and Semigran MJ. Hemodynamic effects of inhaled nitric oxide in right ventricular myocardial infarction and cardiogenic shock. J Am Coll Cardiol 2004; 44: 793-8. [ Links ]
43. Fujita Y, Nishida O, Sobue K, Ito H, Kusama N, Inagaki M, Katsuya H. Nitric oxide inhalation is useful in the management of right ventricular failure caused by myocardial infarction. Crit Care Med 2002; 30 (6): 1379-81. [ Links ]

