










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Cardiología
Print version ISSN 0120-5633
Rev. Col. Cardiol. vol.14 no.4 Bogota July/Aug. 2007
(1) Centro de Investigación e Innovación en Bioingeniería, Universidad Politécnica de Valencia, Valencia, España.
(2) Unidad de Cardiología, Hospital Universitario San Jorge, Universidad Tecnológica de Pereira, Pereira, Colombia.
Correspondencia: Oscar Henao, PhD. Centro de Investigación e Innovación en Bioingeniería, Valencia, España. Correo electrónico: oshegal@doctor.upv.es
Recibido: 31/05/06. Aprobado: 29/03/07.
Las arritmias ventriculares usualmente se presentan como consecuencia de isquemia miocárdica aguda, lo cual causa la mayoría de las muertes súbitas. La heterogeneidad cardiaca tanto funcional (diferente comportamiento eléctrico en los miocitos) como bioquímica (diferentes concentraciones iónicas en isquemia), altera el comportamiento eléctrico del ventrículo y genera dispersión en la repolarización del potencial de acción, por lo que se convierte en sustrato funcional para la generación de arritmias fatales.
En este trabajo se revisan las técnicas experimentales para el estudio y la detección de arritmias por reentrada, y se valida un modelo geométrico de isquemia, en presencia de heterogeneidad eléctrica en la región transmural de la pared ventricular. Para ello se utilizaron simulaciones bidimensionales de un modelo biofísicamente detallado y se obtuvieron reentradas lobulares en diferentes configuraciones de las células M de la pared ventricular.
Palabras clave: arritmias ventriculares, células M, heterogeneidad celular, isquemia regional, pared transmural, reentradas de potencial.
Ventricular arrhythmias usually appear as consequence of acute myocardial ischemia, responsible for most of sudden deaths. Functional (different electrical behavior of myocytes) as well as biochemical (different ionic concentrations during ischemia) cardiac heterogeneity alter the ventricle electric performance and generate dispersion in the action potential repolarization that turns into functional substrate for the generation of fatal arrhythmias.
In this work the experimental techniques for the study and detection of arrhythmias by re-entry are reviewed and a geometrical model of ischemia is validated in the presence of electrical heterogeneity in the trans-mural region of the ventricular wall. For this purpose, bi-dimensional simulations of a detailed biophysical model were used and lobular re-entries in different configurations of the M cells of the ventricular wall were obtained.
Key words: ventricular arrhythmias, M cells, cellular heterogeneity, regional ischemia, transmural wall, potential re-entries.
Heterogeneidad cardiaca
La pared ventricular presenta diferencias estructurales en relación con el tipo de células que la componen. Aunque la distinción entre las tres diferentes clases de células apenas se está aceptando, los trabajos de investigación liderados por Antzelevitch y su grupo no dejan ninguna duda de su existencia (1-3). Las células epicárdicas, las del medio miocardio y las endocárdicas tienen diferencias morfológicas en su potencial de acción, en especial en su particular forma de espiga y domo, localizado en las células M, en el epicardio y casi ausente en las células del endocardio. Esta morfología presente en la fase uno del potencial de acción, se debe a una prominente ranura mediada por la corriente transitoria de salida de potasio (Ito) (4). La forma característica de espiga y domo del potencial, se ha validado de manera experimental utilizando la técnica de fijación de potencial dinámico, que permite inyectar una corriente simulada (Ito) bajo diferentes valores de voltaje fijo, en miocitos dializados de ventrículos caninos (5).
Las células M se pueden distinguir de otros tipos de células debido a que presentan una pequeña corriente rectificadora retardada de potasio (IKs) (6), una larga y tardía corriente de sodio (INa,tardia) (7) y una larga corriente electrogénica del intercambiador sodio-calcio (INa,Ca) (8). La rápida activación del rectificador retardado (IKr) y las corrientes rectificadores de entrada (IK1), son similares a las de los otros tres tipos de células. Estas diferencias iónicas, son responsables del alargamiento en la duración del potencial de acción (APD), y de la gran rapidez en su inicio, la cual es más pronunciada en presencia de fármacos antiarrítmicos de la clase III de Vaughan-Williams. La prolongación preferencial de este potencial de acción en las células M, es causante de una dispersión transmu-ral de repolarización (TDR), la cual puede ser estimada en el electrocardiograma de superficie, como el intervalo entre el pico y el final de la onda T (intervalo QTpico-QTfinal).
Las características histológicas de las células M, son similares a las epicárdicas y las endocárdicas, pero su electrofisiología y su comportamiento farmacológico parece ser un híbrido entre las células de Purkinje y las ventriculares. La células M que presentan los más largos potenciales de acción, a menudo están localizadas en lo profundo de la pared anterior y lateral, y en el tracto de salida del ventrículo derecho entre el subendocardio y el medio miocardio. También existen algunas en los músculos papilares, la trabécula y el septum interventricular (9).
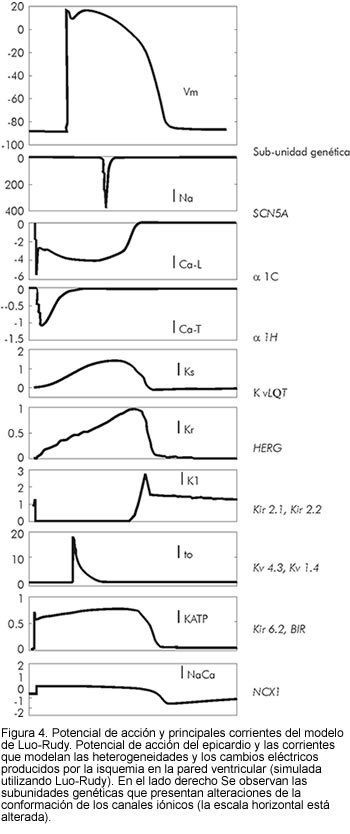
La localización de células M en la pared anterior del ventrículo izquierdo, se halló en lo profundo del subendocardio, según estudios histológicos de Streeter (10) y datos morfológicos y de arquitectura del tejido ventricular revisados por Lunkenheimer (11). En la figura 1 se muestra la comparación de los potenciales de acción correspondientes a la célula intermedia de un tejido 100x100 simulado, utilizando cinética Luo-Rudy y adicionándole heterogeneidad eléctrica (12-14).

Las mayores diferencias entre la magnitud de la ranura del potencial de acción y la (Ito), se han descrito en células M del epicardio ventricular derecho e izquierdo. Las diferencias transmurales e interventriculares en la corriente de salida transitoria tienen interesantes consecuencias como:
- La creación de un gradiente transmural en la manifestación de la ranura del potencial de acción, siendo responsable de la inscripción de la onda J del electrocardiograma (15, 16).
- Sensibilidad diferencial a isquemia y sus respectivos componentes (17).
- Sensibilidad diferencial a neurohormonas, acetilcolina y fármacos tales como isoproterenol, calcioantagonistas, bloqueadores de los canales de sodio/potasio y activadores de los canales de potasio (18, 19).
En el cuadro de la figura 2 se resumen los síndromes generados por TDR que promueven fibrilación ventricular. La heterogénea repolarización del potencial de acción epicárdico, puede ocasionar reentrada de fase dos, la cual produce extrasistolia ventricular capaz de precipitar reentrada de movimiento circular o fibrilación ventricular (Torsade de Pointes rápida).

La amplificación de heterogeneidades intrínsecas en el miocardio ventricular, puede predisponer al desarrollo de arritmias potencialmente letales. Diferentes canalopatías como los síndromes de QTc largo, Brugada y QTc corto, son ejemplo de patologías que presentan fenotipos y etiologías muy diferentes, pero convergen en un camino común causante de la muerte súbita (19). Los desórdenes genéticos responsables de la disfunción en los canales iónicos que manejan la despolarización y repolarización de la membrana, se muestran en la figura 4. Las características clínicas y los cambios en los canales que median la repolarización celular, se resumen en tabla 1 (ver revisiones de Roden (20) y Marban (21)).
La distribución topográfica de las células M en la pared ventricular y su rol funcional en la formación de la taquicardia ventricular polimorfa, variedad Torsade de Pointes, fueron estudiados por Akar (22) quien utilizó mapeos ópticos transmurales en porciones de tejido cardiaco canino, permitiendo una detallada y simultánea medida de la repolarización celular en cada tipo de célula, a través de la pared ventricular, en donde la influencia del acople celular se conocía antes de definir la localización de las células M. Este estudio mostró la primera evidencia experimental sobre la influencia de la distribución topográfica de las células M, en un modelo sucedáneo de la variedad 2 del síndrome de QTc prolongado congénito (LQT2), las cuales causan bloqueo unidireccional y excitación reentrante que lleva a Torsade de Pointes.
La localización topográfica de las células del medio miocardio y su influencia en el acople mecánico en cobayas estudiadas por Wan (23), mostró diferencias transmurales y no longitudinales en la duración del potencial de acción, siendo mayor en el endocardio y en las células M que en el epicardio. Ello sugiere que el gradiente de heterogeneidad en la composición celular de la pared cardiaca, se relaciona de manera inversa con el proceso de despolarización del ventrículo, lo cual contribuye a la morfología de la onda T del electrocardiograma de superficie. Las diferencias regionales en el acortamiento de la amplitud celular, se deben a la variación dinámica en el esfuerzo de la pared, mayor en el endocardio que en el epicardio.
La formación de gradientes transmurales de repolarización entre las distintas células del ventrículo en condiciones normales, parece no ser suficiente para promover la formación de arritmias; sin embargo, el remodelamiento de los canales iónicos y/o de las resistencias asociadas a los espacios intercelulares («gap juntions») en algunas entidades como en la isquemia o en el infarto del miocardio, pueden ser suficientes para incrementar la dispersión de repolarización transmural y de esta forma, el riesgo de arritmias reentrantes. La conductancia unitaria de un canal depende de una familia de proteínas (conexinas) que componen el canal, así las «gap juntions» del músculo ventricular tienen una densidad mayor de conexín 43. La identificación y cuantificación de esta proteína, utilizando inmunofluorescencia, sirve como marcador de la dispersión transmural de repolarización, y de esta forma como detector de los diferentes tipos de células en el ventrículo. Con esta técnica Poelzing (24, 25) logró demostrar que la cantidad de expresión de dicha proteína en el epicardio, es menor que en las células M y en el endocardio, en la dirección de la base al ápex. Además, la conexín 43 se relacionó con la medición de la velocidad de conducción transmural en la pared cardiaca sana, y se encontró un patrón disímil al estimular en epicardio o en endocardio; dicha propagación se asemeja a un frente de onda plano, con una velocidad de propagación de 48 cm/s, si la estimulación es en el endocardio, y tiene forma oblonga con una velocidad de 33 cm/s si la estimulación es en el epicardio (26).
La característica electrofisiológica particular de las células M (su largo APD cuando es activado a longitudes del ciclo cardiaco largo), está muy limitada a células ventriculares aisladas, sin embargo, no es muy clara en la pared ventricular intacta debido al acople de resistencia intercelular y las diferentes interacciones electrotónicas que tienden a ecualizar cualquier diferencia de potencial, yuxtaponiendo largos y cortos potenciales de acción en la pared cardiaca (27). Los efectos combinados de todas las corrientes de membrana y su heterogeneidad en la densidad de canales iónicos, determinan el APD, causando variaciones regionales en su comportamiento. Luego podría ser, que bajo estas condiciones, la diferenciación celular de las células M fuese un estado funcional relacionado con diferentes condiciones de agentes moduladores externos, como el ATX-II y longitudes del ciclo cardiaco variables (28).
Los mecanismos generadores de taquicardia ventricular polimorfa variedad Torsade de pointes, en especial los cambios electrofisiológicos inducidos por pausa en un ciclo corto-largo-corto, pueden acrecentar la dispersión de repolarización y la formación de despolarizaciones tempranas en el ventrículo. Se piensa que la aparición espontánea de esta arritmia, puede deberse al disparo de un latido inducido por despolarizaciones tempranas generadas cercanas al endocardio (en fibras de Purkinje), para luego encontrar un intenso gradiente de repolarización en la región del medio miocardio, bloqueando de manera unidireccional la propagación del potencial y generando así un sustrato funcional para excitación reentrante. Sin embargo, la propagación del potencial ha de ser en la dirección del gradiente de repolarización en donde es más probable que se presente un bloqueo unidireccional; esto sugiere un mecanismo por el cual las células M serían las responsables de la actividad de disparo, induciendo despolarizaciones tempranas y bloqueo unidireccional durante secuencias de longitud de ciclo corto-largo (29).
Isquemia miocárdica aguda
La muerte súbita promovida por fibrilación ventricular, generalmente ocurre durante isquemia miocárdica aguda (30, 31). La interrupción abrupta del flujo de sangre por ruptura de una placa ateromatosa, causa una serie de cambios bioquímicos en la pared del miocardio, lo cual altera la redistribución iónica de sodio, calcio, hidrógeno y potasio a través de la membrana celular (32, 33). Esta serie de eventos, ocurre en los primeros minutos, alterando el comportamiento electrofisiológico de los canales iónicos, y en especial activando los canales de potasio dependientes de ATP (34). Los delicados detalles químicos de interacción mutua en la generación de un latido prematuro y el mantenimiento de arritmia por reentrada en la pared cardiaca, aún están por determinarse. La pérdida de potasio del interior celular y su posterior acumulación en el exterior durante la secuencia hipoxia/isquemia aguda, causan una sostenida despolarización de la membrana, que produce una lenta velocidad de conducción del potencial, generando cambios progresivos en la repolarización, el cual combinado con disminución del pH intracelular y disminución de oxígeno, promueven taquicardia o fibrilación ventricular (35). El bloqueo del metabolismo oxidativo y los cambios en las concentraciones intracelulares de ADP, producen importantes disbalances iónicos, disturbios en el metabolismo lipídico con acumulación de acilcarnitinas, lipofosfoglicéridos, ácidos grasos y ácido araquidónico. Esto lleva a la formación de radicales libres, secreción de neurotransmisores, hormonas y metabolitos con estimulación concomitante de receptores muscarínicos, adrenérgicos y purinérgicos; esto a su vez ocasiona desórdenes mecánicos y de esfuerzo en las regiones isquémicas del tejido (36). El acortamiento del potencial de acción durante la isquemia, se debe a la activación de los canales de potasio sensibles al ATP (IKATP) y al canal de potasio dependiente del ácido araquidónico (IKAA).
La disminución de la energía libre de la hidrólisis del ATP, la acumulación intra y extracelular de H+, Na+ y Ca2+ y los metabolitos lipídicos, afectan de manera directa los receptores del sarcolema, los canales iónicos, los intercambiadores y el potencial de membrana. De manera general, la corriente de excitación es reducida por la acidosis y por la despolarización de la membrana asociada a la acumulación extracelular de K+; se presenta un cambio bifásico en la velocidad de propagación del potencial de acción (37). Todo esto serviría como mecanismo de protección para los miocitos alterados, lo que hace más lenta la conducción eléctrica y se convierte en un sustrato conductivo para la reentrada. En la figura 3 se muestra un esquema de los cambios producidos por isquemia en el tejido miocárdico que son modelados en este trabajo (30, 38).

Las corrientes de repolarización, en particular la corriente de salida de potasio (Ito), la corriente rectificadora retardada (IKr), y la corriente rectificadora de entrada (IK1) son sensibles a los cambios metabólicos del medio. La corriente Ito pierde amplitud en presencia de cambios en la concentración de potasio extracelular y de esta manera la forma de espiga y domo característica del potencial de acción, se vería atenuada, generando una reexcitación en células del epicardio, y ocasionando una extrasístole sobre la onda T, con la consecuente fibrilación ventricular a través de la reentrada de fase dos (39).
La alteración del sodio intracelular en episodios isquémicos del miocardio, es el resultado de dos cambios opuestos: a) La inhibición parcial de la bomba sodio- potasio por hipoxia, déficit de glucosa y acidosis; y b) Una disminución en la entrada pasiva de sodio, seguida por la acumulación de potasio extracelular (33).
Los episodios isquémicos se dividen en tres fases siguiendo la caracterización bifásica del tiempo en relación con la acumulación de potasio (32):
- Primera fase (primeros minutos de oclusión arterial): se caracteriza por rápidos cambios energéticos y acelerada acumulación extracelular de potasio.
- Segunda fase (5 a 20 minutos): presencia de meseta donde tiene lugar un ligero descenso del potasio extracelular.
- Tercera fase: existencia de un abrupto y rápido incremento del potasio extracelular, produciendo una lesión irreversible.
La acumulación de potasio extracelular causa diversos cambios a nivel del tejido; la célula se despolariza y el potencial de acción disminuye su amplitud, incremento y duración. La excitabilidad celular decrece debido a la inactivación en la conductancia del Na, lo cual produce post-repolarizaciones en la refractariedad. Estos cambios en la excitabilidad, refractariedad y conducción, aunados al acortamiento del potencial favorecen la aparición de reentradas (40, 41).
La sensibilidad del epicardio a los cambios bioquímicos generados por eventos isquémicos, ha sido estudiada en modelos experimentales de diversos mamíferos y en corazones humanos. Gilmour, utilizando perfusión con solución Tyrode alterada, que mimetiza los cambios químicos de la isquemia, en porciones de tejido ventricular izquierdo canino, cuantificó la sensibilidad del epicardio (30% de acortamiento del potencial) con respecto al endocardio (10% al 20% de acortamiento) (42).
La reducción en la concentración de ATP, provoca corrientes a través de los canales de potasio, especialmente en las células epicárdicas. La rápida activación de los canales de potasio activados por el ATP en el epicardio, parece ser responsable del acortamiento del potencial en el epicardio, durante la isquemia en felinos (43). En caninos, la activación de los canales de potasio activados por ATP parece involucrarse en el acortamiento de la duración del potencial de acción en isquemia y la elevación del potasio extracelular. Una respuesta diferencial entre la capa epicárdica y la endocárdica ante una menor reducción del ATP intracelular, sugiere un bajo umbral para activación y/o una densa distribución de los canales de KATP en la capa epicárdica (44). Este bajo umbral explicaría, al menos en parte, la supervivencia preferencial de la capa frente a la necrosis durante una prolongada isquemia regional (45, 46).
La elevación de potasio extracelular en la pared cardiaca es heterogénea; aparecen similitudes entre el centro y los márgenes laterales de la zona isquémica en el medio miocardio, entre subendocardio y el subepicardio en el centro de la zona lesionada, y entre electrodos estrechamente separados localizados en el centro de la lesión isquémica en el medio miocardio (47). En roedores, la elevación de potasio extracelular ha sido medida en corazón completo de ratas sometidas a isquemia global y diferentes ambientes gaseosos; dicha elevación de potasio fue más marcada en la parte del medio miocardio, en presencia de solución enriquecida con oxígeno. El gradiente espacial para la variación de potasio se extiende 1 mm a partir del epicardio (48). Estas diferencias en respuesta a hiperkalemia, pueden contribuir en el aplanamiento e inversión de la onda T del ECG (49).
El desbalance en la expresión de la corriente transitoria de potasio (Ito) en epicardio con relación al endocardio, produce un acortamiento del potencial de acción del orden del 60% en el epicardio, con respecto al 20% del endocardio (17), lo cual potencia la influencia de Ito en la dispersión de repolarización y sirve como amplificador de los cambios sobre la excitabilidad producidos por la isquemia en la corriente de calcio, a través de los canales tipo L en los miocitos ventriculares. Bloqueadores específicos de la corriente Ito en miocitos de cobaya y ratones, permitieron establecer la relación funcional entre la corriente transitoria de salida y la corriente de calcio a través de estos canales (50). Las diferencias en depresión de ICa-L en el epicardio versus el endocardio, son un factor más en la respuesta diferencial a eventos isquémicos (51).
La influencia de la acidosis a nivel celular es representada por la disminución del potencial de reposo, la velocidad de despolarización y la prolongación en la duración del potencial de acción (52), con algunas oscilaciones a nivel de la meseta y la ocurrencia de pos-despolarizaciones tempranas (53); todos los cambios inducidos por acidosis son proarrítmicos, lo que se explica por la reducción en el umbral para la fibrilación ventricular (54).
Existen algunos modelos matemáticos de la corriente de potasio dependiente de ATP que permiten caracterizar eventos isquémicos:
- Canal de potasio dependiente de ATP en isquemia, Ferrero (34).
- Canal de potasio dependiente de ATP en isquemia, Shaw-Rudy (55).
- Comportamiento bifásico de potasio extracelular en isquemia, Rodríguez (52).
- Metabolismo de ATP en acople excitación-contracción, Matsuoka (56).
- Canal de potasio dependiente de ATP-ADP libre regulado por magnesio, Michailova (57).
Reentradas asociadas a heterogeneidad ventricular producida por isquemia
La arritmia por reentrada es el resultado de un disturbio en la propagación del potencial, que no cesa después de una activación completa de las aurículas y los ventrículos, dejando reexcitado el tejido, después del final del periodo refractario (58). Para que se presenten arritmias por reentradas en el ventrículo deben ocurrir simultáneamente dos condiciones esenciales:
a) La existencia de un bloqueo unidireccional en la propagación del impulso en cierto lugar del camino (ventana vulnerable) (59) de conducción a través de la pared ventricular.
b) La combinación de la conducción lenta y un corto periodo refractario en la parte remanente del camino de conducción (60).
En isquemia se logran ambas condiciones; debido a la hiperkalemia, en los primeros instantes del bloqueo de flujo sanguíneo, la despolarización de la membrana bloquea los canales de sodio y decrece la corriente generada durante el incremento del potencial de acción. La velocidad de conducción decrece, el acople intercelular disminuye y se acorta el potencial de acción, permitiendo un alargamiento en el periodo refractario efectivo. La refractariedad no es homogénea y la conducción es anisotrópica y reducida (61) específicamente en la zona de borde que separa al tejido isquémico de la zona sana. Todos estos cambios no ocurren de manera uniforme en la zona afectada por la lesión, la heterogeneidad es más pronunciada en la zona borde, comparada con la zona que limita la parte enferma del miocardio (62).
Reentrada de fase dos
La presencia de una corriente adicional de salida Ito en el epicardio ventricular, predispone la pérdida del domo en el potencial de acción durante la isquemia, lo cual permite la amplificación de las heterogeneidades transmurales y el desarrollo de reentrada de fase dos y taquicardia. Las arritmias ventriculares tempranas, durante los primeros minutos de isquemia miocárdica, ocurren en una distribución temporal bifásica, durante la fase 1a (inmediata) y la fase 1b (retardada). En la fase 1a la fibrilación ocurre en el intervalo comprendido entre los 2 y 10 minutos desde el comienzo de la oclusión coronaria, con un pico de incidencia entre el minuto 5 y 6, cuando la concentración de potasio extracelular se acerca a su fase de meseta (63). La pérdida del domo mediado por Ito del potencial de acción epicárdico en la fase 1a de la isquemia aguda y en la zona de borde, genera reentrada de fase 2, que se manifiesta como una extrasístole acoplada de R sobre T en el electrocardiograma, como lo demostró Yan (64) de manera experimental en preparaciones caninas de pared ventricular y Miyoshi (65) en preparaciones simuladas utilizando cinética Luo-Rudy.
Reentrada en forma de ocho
Los trabajos de El-Sherif (66-69) describieron por primera vez esta reentrada en capas de epicardio, que sobrevivió a infarto severo después de la oclusión de la arteria descendente anterior en corazones caninos. En el modelo de figura en ocho, el latido reentrante genera un frente de onda que circula en torno a una línea de bloqueo funcional, uniéndose en la parte distal del bloqueo. El singular arco de bloqueo es dividido en dos partes y la activación reentrante continúa como dos frentes de onda circulantes que viajan a favor y en contra del movimiento de las manecillas del reloj, en una configuración tipo ocho horizontal. Este tipo de reentrada, se ha reportado en corazones porcinos utilizando mapeos eléctricos de superficie, en la primera fase de isquemia aguda (70). Esta arritmia la estudió de manera teórica Ferrero, mediante simulación bidimensional, utilizando cinética Luo-Rudy en un modelo de isquemia regional aguda (71).
Reentrada espiral
El término espiral, introducido por Wiener (72), se utiliza para describir la rotación de un frente de onda de potencial en torno a un bloqueo funcional; Winfree desarrolló este concepto de manera extensa (73). La curvatura de la espiral es el concepto clave para la formación de la singularidad de fase, asociada a una región donde en un punto convergen el frente de onda y su cola refractaria, formando una desestabilización de tipo fuente-sumidero. La corriente provista por el frente reentrante (fuente), es insuficiente para cargar la capacidad de membrana, excitando volúmenes de tejido cuyo periodo refractario no ha terminado (sumidero) (74, 75). Este tipo de arritmias se ha encontrado en porciones transmurales de tejido cardiaco canino, utilizando mapeos ópticos en preparaciones perfundidas tipo Langerdorff (76, 77).
Caso de estudio
Estamos interesados en estudiar los diferentes mecanismos iónicos generadores de patrones reentrantes en isquemia aguda, en la pared transmural cardiaca. Los diferentes componentes de isquemia: hipoxia, acidosis e hiperkalemia han sido modelados e incluidos en la descripción cinética de iones a través de la membrana, desarrollada por Luo-Rudy (13, 14, 78). La heterogeneidad cardiaca es incluida en el modelo por medio de la corriente transitoria de salida Ito (12) y la componente lenta del rectificador de potasio IKs. Los valores de las conductancias de estas corrientes fueron alterados por defecto, para modelar los tres diferentes tipos de células que componen la pared transmural (79).
Materiales y métodos
La dinámica del modelo describe las principales corrientes iónicas responsables de la forma de onda del potencial. El modelo incluye 16 diferentes corrientes iónicas a través del sarcolema (incluyendo distintas corrientes de Na+, K+ y Ca2+, así como también bombas electrogénicas e intercambiadores), el vaciado de Ca2+ inducido por Ca2+ y almacenes de Ca2+ intracelular. La estructura matemática básica de una célula cardiaca del modelo, está gobernada por la ecuación de reacción-difusión (80):

donde Ic es la corriente a través de la membrana, Cm es la capacitancia de la membrana celular, Vm es el potencial, Iion=(Ib+Ii+Is) es la suma de las corrientes por unidad de área en la descripción de Luo-Rudy y Iest es el estímulo de corriente aplicado, o es la conductividad del tejido, en este caso tomada como lineal anisotrópica. Cada corriente a través de un canal específico es descrita por:
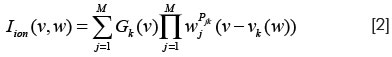
donde Vk(w) es el potencial de Nernst dependiente de la concentración de un ion k específico, Gk(V) es la conductancia del canal iónico especificado por k, M son las compuertas, w:(w1,...,WM) las concentraciones y los Pjk son enteros. La conductancia de un canal es descrita por una ecuación diferencial no lineal siguiendo el formalismo de Hodgkin-Huxley (81):
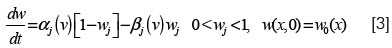
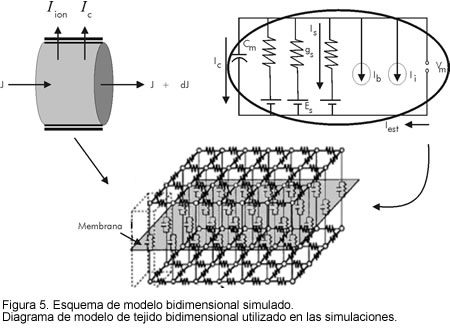
En la figura 5 se muestra un diagrama esquemático del tejido bidimensional simulado. La parte superior izquierda explica gráficamente la propagación del potencial a través del tejido (lado izquierdo de la ecuación [1]) y la parte derecha muestra el circuito de membrana equivalente, utilizado para modelar las propiedades activas del tejido (parte derecha de la ecuación [1]).
La parte superior izquierda de la figura 6, muestra la estructura electrofisiológica del tejido modelado (correspondiente al minuto ocho después del comienzo de la isquemia), en el cual existen diferentes zonas que representan los cambios generados por la isquemia. El tamaño de estas zonas, así como los valores relevantes de la concentración extracelular de potasio y sus gradientes espaciales, se obtuvieron de datos experimentales revisados por Coronel (35) y de datos de simulación (52). Una zona central (CZ) de forma semicircular representa las células con falta de flujo sanguíneo, una zona también semicircular correspondiente con la zona de borde (BZ) del tejido en la cual los parámetros de hipoxia, acidosis e hiperkalemia del modelo cambian espacialmente, y una zona normal de la pared ventricular (NZ) rodeando la zona de borde. Los valores de los parámetros que modelan la acidosis y la hiperkalemia en la lesión isquémica fueron descritos anteriormente por Ferrero (71). Se sabe que en la hipoxia decrece la entrada de oxígeno después de la oclusión de la arteria coronaria lo que lleva a disminuir la concentración intracelular de ATP y a aumentar la concentración de ADP. Estos cambios afectan la activación de la corriente de potasio dependiente de ATP (IKATP), la cual permanece casi inactiva en miocardio normal. Se usó un modelo matemático de IKATP desarrollado en nuestro laboratorio, el cual considera que la corriente depende de las concentraciones de ATP, ADP, Mg+ intracelulares y de la concentración extracelular de K+, como también de la dependencia de los canales con respecto al voltaje de membrana. La corriente y la fracción de canales abiertos considerados por el modelo se suscriben como:
los niveles de ATP y ADP proporcionan una fracción de canales dada por fATP=0% y fATP=0.6% en la NZ y la CZ respectivamente (34).

Las diferentes configuraciones de células M, se basan en trabajos experimentales con porciones de tejido ventricular canino (22, 82) su distribución es en aglomerado alargado, aglomerado circular en la zona media, y sin aglomerado de células M, en donde el epicardio ocupa el 20% del tejido, como se muestra en la parte superior derecha de la figura 6. El protocolo S1-S1 de estimulación del tejido, sigue el esquema de un primer estímulo (intensidad doble de umbral diastólico de 2 ms de duración) aplicado en la parte superior del tejido modelado 50 ms después del comienzo de la simulación, y un segundo estímulo prematuro aplicado en la parte refractaria del primer potencial generado en el mismo lugar del tejido, con el fin de lograr la interacción de frente de onda y cola refractaria necesaria para la reentrada. El instante temporal de aplicación del segundo estímulo se realiza con un intervalo de acople (CI) de 180 ms, ubicado dentro de la ventana vulnerable del tejido (83). El diagrama de estimulación se muestra en la esquina inferior de la figura 6.
Resultados
En la figura 7 aparecen fotogramas de las simulaciones realizadas en diferentes tiempos. De izquierda a derecha y en forma vertical, se obtiene el progreso del primer estímulo (80 ms), y el segundo impulso en el tercer fotograma (265 ms). En el cuarto fotograma (300 ms), se muestra el bloqueo en la zona de borde isquémico y la formación del patrón de potencial reentrante en torno a la lesión simulada; los siguientes fotogramas muestran la evolución del circuito reentrante, que gira de manera contraria a las manecillas del reloj, rotando una vez y media alrededor del tejido deprimido por la isquemia. En la parte A de la figura 7, se muestra la reentrada, utilizando una configuración en banda alargada de células M, y en la parte B una reentrada con distribución circular de las mismas células. Se puede notar el retraso en la velocidad del frente de onda y el cambio en la morfología del mismo frente, al interactuar con las células M.
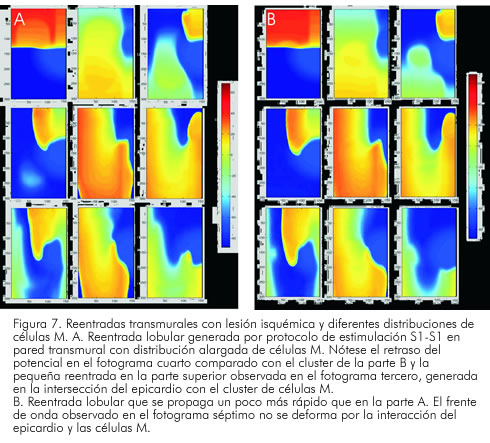
En el minuto ocho de isquemia estudiado, el estímulo prematuro se bloquea en el endocardio y presenta un retraso que le da una forma de horquilla en la propagación del frente. Se produce un bloqueo unidireccional en la zona de borde, formándose un núcleo que rota una vez y media en torno a la zona isquémica, para luego bloquearse de manera bidireccional en la parte inferior de la lesión y difuminarse. El núcleo del rotor permanece en la región isquémica (Figura 8). Una observación importante es la gran sensibilidad del epicardio ante los cambios modelados de la isquemia; la heterogeneidad en la excitabilidad del tejido simulado para esta estimulación, provee las condiciones necesarias para la generación, el mantenimiento y la terminación autónoma de los frentes reentrantes (77). La isquemia aguda modelada aumenta la dispersión transmural, con mayor depresión de excitabilidad y conducción en el epicardio que en el endocardio. Esta respuesta diferencial, permite que la activación iniciada por el estímulo prematuro en el epicardio, se bloquee en el borde de la lesión en la parte del epicardio, despolarizando de manera lateral la zona de borde externa del endocardio, y retorne al epicardio para formar el lazo reentrante. Este disbalance en la excitación y la conducción del frente de onda, crea el sustrato para generar el patrón reentrante observado. Las respuestas diferenciales del epicardio y las células M, proveen sustrato para bloqueo unidireccional, creando un camino funcional para la generación de reentradas durante isquemia, y se establece una posible «ventana de oportunidad» para eventos que desestabilizan el frente de onda.

Discusión
La utilización de modelos computacionales para evaluar la propagación eléctrica cardiaca, permite la disponibilidad de numerosas variables que no están disponibles en ensayos experimentales tanto in vitro como in vivo. La validación de este modelo de propagación, utilizando propiedades activas de membrana, la ha llevado a cabo nuestro grupo en isquemia en tejidos homogéneos, modelo de fármacos, modelo de acumulación de potasio extracelular y formación de despolarizaciones tempranas al variar la resistencia intercelular (52, 71, 84, 85).
Las simulaciones bidimensionales de tejido isquémico en la pared heterogénea ventricular son difíciles de lograr. Existen algunos reportes de simulaciones bidimensionales que utilizan cinética Luo-Rudy, combinada con cadenas de Markov que describen la corriente de sodio para estudiar la desestabilización de ondas espirales en síndromes de QTc prolongado, con heterogeneidad manejada por la corriente retardada de potasio (86). Estos resultados podrían confrontarse con los nuestros y comparar la desestabilización del frente de onda, pues su mecanismo obedece a la variación de conductancias en la corriente retardada de potasio; el nuestro es una variación de los parámetros electroquímicos que modelan los cambios en isquemia regional, presentando diferencias en los frentes de onda reentrante. Un rotor en torno a la lesión isquémica, se difumina después de algunas vueltas en torno al núcleo, ocasionando desestabilización del frente, secundaria a la dispersión de repolarización manejada por las corrientes salientes de potasio.
La simulación en tejido bidimensional para rotores de potencial, con estimulación en presencia de gradientes de potencial que pueden detener el frente reentrante, fue llevada a cabo por Yao (87), en tejidos bidimensionales, emulando comportamiento en dos dominios de la pared. Sus resultados detienen el frente de onda anisotrópico y reestablecen la conducción cardiaca entre diferentes puntos del tejido. El frente reentrante hallado en las simulaciones realizadas, se detiene de manera autónoma por el tejido y la dispersión en la velocidad de conducción entre zona sana y lesionada, y crea el bloqueo unidireccional en la parte proximal de la lesión. La detención del frente de onda en la parte distal del tejido, es consecuencia de un bloqueo bidireccional generado por dispersión del periodo refractario, al encontrarse el frente con una zona que todavía se halla en periodo refractario relativo y por tanto inexcitable. Este tipo de frente de onda reentrante, que se propaga desde el epicardio hacia el endocardio, fue obtenido de manera experimental por Zipes (76) en porciones de pared ventricular izquierda de corazón canino, bajo condiciones simuladas de isquemia global, pero con un esquema de estimulación diferente.
Valderrabano (88, 89) observó la reentrada en la región transmural de la pared, en porciones de ventrículos de corazón de cerdo; el rotor hallado gira dentro de la pared y luego se difumina en el tejido, mostrando cómo la anisotropía del mismo es fundamental para el mantenimiento del patrón. En este estudio, se modela anisotropía funcional generada por isquemia y por la interfase epicardio-endocardio que desestabiliza el frente y promueve la reentrada en la porción intramural simulada. Las reentradas obtenidas muestran la sensibilidad del epicardio, ante alteraciones bioquímicas generadas por la isquemia. La zona de borde bloquea el frente de manera funcional y por despolarización transversal del tejido circundante, genera la formación del frente reentrante.
La dispersión en la velocidad de conducción y la alteración del delicado balance entre duración del potencial de acción y el periodo refractario celular, crean un sustrato para la formación y terminación autónoma de reentradas de potencial, en presencia de isquemia transmural. Estos resultados pueden considerarse como un ingrediente adicional en la generación de fibrilación ventricular, y concuerdan con los hallazgos experimentales de fibrilación ventricular de eventos isquémicos (90, 91).
Conclusión
El modelo desarrollado en este trabajo, permite estudiar la amplificación de heterogeneidades transmurales de repolarización del potencial de acción, causadas por la isquemia, las diferentes configuraciones de células M modeladas y su influencia en el potencial desarrollo de arritmias reentrantes letales. Los síndromes de QT largo, QT corto, Brugada y taquicardia ventricular por catecolaminas, son canalopatías con muy diferentes fenotipos y etiologías, pero que convergen en la dispersión de repolarización, que puede estudiarse con el modelo desarrollado. Aunque el modelo es una simplificación de la gran cantidad de eventos bioquímicos generados en isquemia en la pared transmural, permite acercarse cada vez más, desde el punto de vista teórico, al entendimiento de los complejos mecanismos que suscitan fibrilación ventricular, y que conllevan muerte súbita.
Agradecimientos
Este trabajo fue parcialmente financiado por el Plan Nacional de Investigación Científica, Desarrollo e Innovación Tecnológica del Ministerio de Educación y Ciencia (proyectos TIN2004-03602 y TEC 2005-04199/TCM), de España.
Bibliografía
1. Antzelevitch C, Fish J. Electrical heterogeneity within the ventricular wall. Basic Res Cardiol 2001 96; 2001 (6): 517-527. [ Links ]
2. Antzelevitch C, Sicouri S, Litovsky SH, Lukas A, Krishnan SC, Di Diego JM, et al. Heterogeneity within the ventricular wall. Electrophysiology and pharmacology of epicardial, endocardial, and M cells. Circ Res 1991; 69 (6): 1427-1449. [ Links ]
3. Sicouri S, Antzelevitch C. A subpopulation of cells with unique electrophysiological properties in the deep subepicardium of the canine ventricle. The M cell. Circ Res 1991; 68 (6): 1729-1741. [ Links ]
4. Liu DW, Gintant GA, Antzelevitch C. Ionic bases for electrophysiological distinctions among epicardial, midmyocardial, and endocardial myocytes from the free wall of the canine left ventricle. Circ Res 1993; 72 (3): 671-68. [ Links ]
5. Sun X, Wang HS. Role of the transient outward current (Ito) in shaping canine ventricular action potential—a dynamic clamp study. J Physiol 2005; 564 (Pt 2): 411-419. [ Links ]
6. Liu DW, Antzelevitch C. Characteristics of the delayed rectifier current (IKr and IKs) in canine ventricular epicardial, midmyocardial, and endocardial myocytes. A weaker IKs contributes to the longer action potential of the M cell. Circ Res 1995; 76 (3): 351-365. [ Links ]
7. Zygmunt AC, Eddlestone GT, Thomas GP, Nesterenko VV, Antzelevitch C. Larger late sodium conductance in M cells contributes to electrical heterogeneity in canine ventricle. Am J Physiol Heart Circ Physiol 2201; 281 (2): p. H689-H697. [ Links ]
8. Zygmunt AC, Goodrow RJ, Antzelevitch C. I(NaCa) contributes to electrical heterogeneity within the canine ventricle. Am J Physiol Heart Circ Physiol 2000; 278 (5): H1671-H1678. [ Links ]
9. Antzelevitch C. Heterogeneity of cellular repolarization in LQTS: the role of M cells. Eur Heart J Suppl 2001; 3 (K): K2-K16. [ Links ]
10. Streeter DD, Jr, Spotnitz HM, Patel DP, Ross J, Jr, Sonnenblick EH. Fiber orientation in the canine left ventricle during diastole and systole. Circ Res 1969; 24 (3): 339-347. [ Links ]
11. Lunkenheimer PP, Redmann K, Anderson RH. The architecture of the ventricular mass and its functional implications for organ-preserving surgery. Eur J Cardiothorac Surg 2005; 27 (2): 183-190. [ Links ]
12. Dumaine R, Towbin JA, Brugada P, Vatta M, Nesterenko DV, Nesterenko VV, et al. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res 1999; 85 (9): 803-809. [ Links ]
13. Luo CH, Rudy Y. A dynamic model of the cardiac ventricular action potential. I. Simulations of ionic currents and concentration changes. Circ Res 1994; 74 (6): 1071-1096. [ Links ]
14. Viswanathan PC, Shaw RM, Rudy Y. Effects of IKr and IKs heterogeneity on action potential duration and its rate dependence: a simulation study. Circulation 1999; 99 (18): 2466-2474. [ Links ]
15. Antzelevitch C, Shimizu W, Yan GX, Sicouri S, Weissenburger J, Nesterenko VV, et al. The M cell: Its contribution to the ECG and to normal and abnormal electrical function of the heart. J Cardiovasc Electrophysiol 1999; 10 (8): 1124-1152. [ Links ]
16. Antzelevitch C. Cellular basis for the repolarization waves of the ECG. Ann N Y Acad Sci 2006; 1080: 268-281. [ Links ]
17. Lukas A, Antzelevitch C. Differences in the electrophysiological response of canine ventricular epicardium and endocardium to ischemia. Role of the transient outward current. Circulation 1993; 88 (6): 2903-2915. [ Links ]
18. Wolk R, Cobbe SM, Hicks MN, Kane KA. Functional, structural, and dynamic basis of electrical heterogeneity in healthy and diseased cardiac muscle: implications for arrhythmogenesis and anti-arrhythmic drug therapy. Pharmacol Ther 1999; 84 (2): 207-231. [ Links ]
19. Antzelevitch C, Oliva A. Amplification of spatial dispersion of repolarization underlies sudden cardiac death associated with catecholaminergic polymorphic VT, long QT, short QT and Brugada syndromes. J Intern Med 2006; 259 (1): 48-58. [ Links ]
20. Roden DM, Balser JR, George AL, Jr., Anderson ME. Cardiac ion channels. Ann Rev Physiol 2002; 64: 431-475. [ Links ]
21. Marban E. Cardiac channelopathies. Nature 2002; 415 (6868): 213-218. [ Links ]
22. Akar FG, Yan GX, Antzelevitch C, Rosenbaum DS. Unique topographical distribution of M cells underlies reentrant mechanism of torsade de pointes in the long-QT syndrome. Circulation 2002; 105 (10): 1247-1253. [ Links ]
23. Wan X, Bryant SM, Hart G. A topographical study of mechanical and electrical properties of single myocytes isolated from normal guinea-pig ventricular muscle. J Anat 2003; 202 (6): 525-536. [ Links ]
24. Poelzing S, Rosenbaum DS. Nature, significance, and mechanisms of electrical heterogeneities in ventricle. Anat Rec A Discov Mol Cell Evol Biol 2004; 280 (2): 1010-1017. [ Links ]
25. Poelzing S, Akar FG, Baron E, Rosenbaum DS. Heterogeneous connexin43 expression produces electrophysiological heterogeneities across ventricular wall. Am J Physiol Heart Circ Physiol 2004; 286 (5): H2001-H2009. [ Links ]
26. Poelzing S, Dikshteyn M, Rosenbaum DS. Transmural conduction is not a two-way street. J Cardiovasc Electrophysiol 2005; 16 (4): 455. [ Links ]
27. Taggart P, Sutton P, Opthof T, Coronel R, Kallis P. Electrotonic cancellation of transmural electrical gradients in the left ventricle in man. Prog Biophys Mol Biol 2003; 82 (1-3): 243-254. [ Links ]
28. Ueda N, Zipes DP, Wu J. Functional and transmural modulation of M cell behavior in canine ventricular wall. Am J Physiol Heart Circ Physiol 2004; 287 (6): H2569-H2575. [ Links ]
29. Liu J, Laurita KR. The mechanism of pause-induced torsade de pointes in long QT syndrome. J Cardiovasc Electrophysiol 2005; 16 (9): 981-987. [ Links ]
30. Rubart M, Zipes DP. Mechanisms of sudden cardiac death. J Clin Invest 2005115 (9): 2305-2315. [ Links ]
31. Zipes DP, Wellens HJ. Sudden cardiac death. Circulation 1998; 98 (21): 2334-2351. [ Links ]
32. Coronel R, Fiolet JW, Wilms-Schopman FJ, Schaapherder AF, Johnson TA, Gettes LS, et al. Distribution of extracellular potassium and its relation to electrophysiologic changes during acute myocardial ischemia in the isolated perfused porcine heart. Circulation 1988 (5): 1125-1138. [ Links ]
33. Wilde AA, Kleber AG. The combined effects of hypoxia, high K+, and acidosis on the intracellular sodium activity and resting potential in guinea pig papillary muscle. Circ Res 1986; 58 (2): 249-256. [ Links ]
34. Ferrero JM, Jr., Saiz J, Ferrero JM, Thakor NV. Simulation of action potentials from metabolically impaired cardiac myocytes. Role of ATP-sensitive K+ current. Circ Res 1996; 79 (2): 208-221. [ Links ]
35. Coronel R. Heterogeneity in extracellular potassium concentration during early myocardial ischaemia and reperfusion: implications for arrhythmogenesis. Cardiovasc Res 1994; 28 (6): 770-777. [ Links ]
36. Carmeliet E. Cardiac ionic currents and acute ischemia: from channels to arrhythmias. Physiol Rev 1999; 79 (3): 917-1017. [ Links ]
37. Kagiyama Y, Hill JL, Gettes LS. Interaction of acidosis and increased extracellular potassium on action potential characteristics and conduction in guinea pig ventricular muscle. Circ Res 1982; 51 (5): 614-623. [ Links ]
38. Cascio WE. Myocardial ischemia: what factors determine arrhythmogenesis? J Cardiovasc Electrophysiol 2001; 12 (6): 726-729. [ Links ]
39. Yan GX, Kowey PR. ST segment elevation and sudden cardiac death: from the Brugada syndrome to acute myocardial ischemia. J Cardiovasc Electrophysiol 2000; 11 (12): 1330-1332. [ Links ]
40. Cascio WE, Johnson TA, Gettes LS. Electrophysiologic changes in ischemic ventricular myocardium: I. Influence of ionic, metabolic, and energetic changes. J Cardiovasc Electrophysiol 1995; 6 (11): 1039-1062. [ Links ]
41. Gettes LS, Cascio WE. Effect of acute ischemia on cardiac electrophysiology. In: The Heart and Cardiovascular System. H. A. Fozzard, E. Haber, R. Jennings, A. Katz, and H. Morgan, Eds. New York: Raven Press ltd.; 1992. p. 2021-2053. [ Links ]
42. Gilmour RF, Jr., Zipes DP. Different electrophysiological responses of canine endocardium and epicardium to combined hyperkalemia, hypoxia, and acidosis. Circ Res 1980; 46 (6): 814-825. [ Links ]
43. Furukawa T, Kimura S, Furukawa N, Bassett AL, Myerburg RJ. Role of cardiac ATP-regulated potassium channels in differential responses of endocardial and epicardial cells to ischemia. Circ Res 1991; 68 (6): 1693-1702. [ Links ]
44. Miyoshi S, Miyazaki T, Moritani K, Ogawa S. Different responses of epicardium and endocardium to KATP channel modulators during regional ischemia. Am J Physiol 1996; 271 (1 Pt 2): H140-H147. [ Links ]
45. Di Diego JM, Antzelevitch C. Pinacidil-induced electrical heterogeneity and extrasystolic activity in canine ventricular tissues. Does activation of ATP-regulated potassium current promote phase 2 reentry? Circulation 1993; 88 (3): 1177-1189. [ Links ]
46. Lowe JE, Cummings RG, Adams DH, Hull-Ryde EA. Evidence that ischemic cell death begins in the subendocardium independent of variations in collateral flow or wall tension. Circulation 1983; 68 (1): 190-202. [ Links ]
47. Hill JL, Gettes LS. Effect of acute coronary artery occlusion on local myocardial extracellular K+ activity in swine. Circulation 1980; 61 (4): 768-778. [ Links ]
48. Schaapherder AF, Schumacher CA, Coronel R, Fiolet JW. Transmural inhomogeneity of extracellular (K+) and pH and myocardial energy metabolism in the isolated rat heart during acute global ischemia; dependence on gaseous environment. Basic Res Cardiol 1990; 85 (1): 33-44. [ Links ]
49. Wan X, Bryant SM, Hart G. The effects of (K+)o on regional differences in electrical characteristics of ventricular myocytes in guinea-pig. Exp Physiol 2000; 85 (6): 769-774. [ Links ]
50. Wang Y, Cheng J, Tandan S, Jiang M, McCloskey DT, Hill JA. Transient-outward K+ channel inhibition facilitates L-type Ca2+ current in heart. J Cardiovasc Electrophysiol 2006; 17 (3): 298-304. [ Links ]
51. Kimura S, Bassett AL, Furukawa T, Furukawa N, Myerburg RJ. Differences in the effect of metabolic inhibition on action potentials and calcium currents in endocardial and epicardial cells. Circulation 1991; 84 (2): 768-777. [ Links ]
52. Rodriguez B, Ferrero JM, Jr., Trenor B.Mechanistic investigation of extracellular K+ accumulation during acute myocardial ischemia: a simulation study. Am J Physiol Heart Circ Physiol 2002; 283 (2): H490-H500. [ Links ]
53. Sato R, Noma A, Kurachi Y, Irisawa H. Effects of intracellular acidification on membrane currents in ventricular cells of the guinea pig. Circ Res 1985; 57 (4): 553-561. [ Links ]
54. Surawicz B. Ventricular fibrillation. J Am Coll Cardiol 1985; 5 (6 Suppl): 43B-54B. [ Links ]
55. Shaw RM, Rudy Y. Electrophysiologic effects of acute myocardial ischemia: a theoretical study of altered cell excitability and action potential duration. Cardiovasc Res 1997; 35 (2): 256-272. [ Links ]
56. Matsuoka S, Sarai N, Jo H, Noma A. Simulation of ATP metabolism in cardiac excitation-contraction coupling. Prog Biophys Mol Biol 2004; 85 (2-3): 279-299. [ Links ]
57. Michailova A, Saucerman J, Belik ME, McCulloch AD. Modeling regulation of cardiac KATP and L-type Ca2+ currents by ATP, ADP, and Mg2+. Biophys J 2005; 88 (3): 2234-2249. [ Links ]
58. Mines G. On dynamic equilibrium in the heart. J Physiol (London) 1913; 46: 349-383. [ Links ]
59. Chen PS, Wolf PD, Dixon EG, Danieley ND, Frazier DW, Smith WM, Ideker RE. Mechanism of ventricular vulnerability to single premature stimuli in open-chest dogs. Circ Res 1988; 62 (6): 1191-1209. [ Links ]
60. Frazier DW, Wolf PD, Wharton JM, Tang AS, Smith WM, Ideker RE. Stimulus-induced critical point. Mechanism for electrical initiation of reentry in normal canine myocardium. J Clin Invest 1989; 83 (3): 1039-1052. [ Links ]
61. Kleber AG, Janse MJ, Wilms-Schopmann FJ, Wilde AA, Coronel R. Changes in conduction velocity during acute ischemia in ventricular myocardium of the isolated porcine heart. Circulation 1986; 73 (1): 189-198. [ Links ]
62. Wilensky RL, Tranum-Jensen J, Coronel R, Wilde AA, Fiolet JW, Janse MJ. The subendocardial border zone during acute ischemia of the rabbit heart: an electrophysiologic, metabolic, and morphologic correlative study. Circulation 1986; 74 (5): 1137-1146. [ Links ]
63. Punske BB, Cascio WE, Engle C, Nagle HT, Gettes LS, Johnson TA. Quantitative characterization of epicardial wave fronts during regional ischemia and elevated extracellular potassium ion concentration. Ann Biomed Eng 1998; 26 (6): 1010-1021. [ Links ]
64. Yan GX, Joshi A, Guo D, Hlaing T, Martin J, Xu X, Kowey PR. Phase 2 reentry as a trigger to initiate ventricular fibrillation during early acute myocardial ischemia. Circulation 2004; 110 (9): 1036-1041. [ Links ]
65. Miyoshi S, Mitamura H, Fujikura K, Fukuda Y, Tanimoto K, Hagiwara Y, et al. A mathematical model of phase 2 reentry: role of L-type Ca current. Am J Physiol Heart Circ Physiol 2003; 284 (4): H1285-H1294. [ Links ]
66. El-Sherif N, Smith RA, Evans K. Canine ventricular arrhythmias in the late myocardial infarction period. Epicardial mapping of reentrant circuits. Circ Res 1981; 49 (1): 255-265. [ Links ]
67. El-Sherif N. Reentry revisited. Pacing Clin Electrophysiol 1988; 11 (9): 1358-1368. [ Links ]
68. Gough WB, Mehra R, Restivo M, Zeiler RH, El-Sherif N. Reentrant ventricular arrhythmias in the late myocardial infarction period in the dog. Correlation of activation and refractory maps. Circ Res 1985; 57 (3): 432-442. [ Links ]
69. Mehra R, Zeiler RH, Gough WB, El-Sherif N. Reentrant ventricular arrhythmias in the late myocardial infarction period. Electrophysiologic-anatomic correlation of reentrant circuits. Circulation 1983; 67 (1): 11-24. [ Links ]
70. Janse MJ, Wit AL. Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischemia and infarction. Physiol Rev 1989; 69 (4): 1049-1169. [ Links ]
71. Ferrero JM, Jr., Trenor B, Rodriguez B, Saiz J. Electrical activity and reentry during acute regional myocardial ischemia: insights from simulations. Intern J Bifurcation Chaos 2003; 13 (12): 3703-3715. [ Links ]
72. Wiener N, Rosenblueth A. The mathematical formulation of the problem of conduction of impulses in a network of connected excitable elements,specifically in cardiac muscle. Archivos del instituto de Cardiología de México 1946; 16 (3,4): 205-265. [ Links ]
73. Winfree AT. The geometry of biological time. 2nd. ed. Springer Verlag; 2001. p. 455-526.
74. Winfree AT. Heart muscle as a reaction-diffusion medium: The roles of electric potential diffusion, activation front curvature, and anisotropy. Intern J Bifurcation Chaos 1997; 7 (3): 487-526. [ Links ]
75. Fast VG, Kleber AG. Role of wavefront curvature in propagation of cardiac impulse. Cardiovasc Res 1997; 33 (2): 258-271. [ Links ]
76. Wu J, Zipes DP. Transmural reentry during acute global ischemia and reperfusion in canine ventricular muscle. Am J Physiol Heart Circ Physiol 2001; 280 (6): H2717-H2725. [ Links ]
77. Wu J, Zipes DP. Transmural reentry triggered by epicardial stimulation during acute ischemia in canine ventricular muscle. Am J Physiol Heart Circ Physiol 2002; 283 (5): H2004-H2011. [ Links ]
78. Faber GM, Rudy Y. Action potential and contractility changes in (Na(+))(i) overloaded cardiac myocytes: a simulation study. Biophys J 2000; 78 (5): 2392-2404. [ Links ]
79. Clayton RH, Holden AV. Dispersion of cardiac action potential duration and the initiation of re-entry: a computational study. Biomed Eng Online 2005; 4 (1): 11. [ Links ]
80. Weidmann S. Electrical constants of trabecular muscle from mammalian heart. J Physiol 1970; 210 (4): 1041-1054. [ Links ]
81. Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol 1952; 117 (4): 500-544. [ Links ]
82. Yan GX, Shimizu W, Antzelevitch C. Characteristics and distribution of M cells in arterially perfused canine left ventricular wedge preparations. Circulation 1998; 98 (18): 1921-1927. [ Links ]
83. Winfree AT. Electrical instability in cardiac muscle: phase singularities and rotors. J Theor Biol 1989; 138 (3): 353-405. [ Links ]
84. Saiz J, Ferrero JM, Jr., Monserrat M, Ferrero JM, Thakor NV. Influence of electrical coupling on early afterdepolarizations in ventricular myocytes. IEEE Trans Biomed Eng 1999; 46 (2): 138-147. [ Links ]
85. Trenor B, Ferrero JM, Jr., Rodriguez B, Montilla F. Effects of pinacidil on reentrant arrhythmias generated during acute regional ischemia: a simulation study. Ann Biomed Eng 2005; 33 (7): 897-906. [ Links ]
86. Henry H, Rappel WJ. The role of M cells and the long QT syndrome in cardiac arrhythmias: simulation studies of reentrant excitations using a detailed electrophysiological model. Chaos 2004; 14 (1): 172-182. [ Links ]
87. Yao T, Ashihara T, Ito M, Nakazawa K, Horie M. Refractory gradient is responsible for the increase in ventricular vulnerability under sodium channel blockade. Circ J 2005; 69 (3): 345-353. [ Links ]
88. Valderrabano M, Lee MH, Ohara T, Lai AC, Fishbein MC, Lin SF, et al. Dynamics of intramural and transmural reentry during ventricular fibrillation in isolated swine ventricles. Circ Res 2001; 88 (8): 839-848. [ Links ]
89. Valderrabano M, Chen PS, Lin SF. Spatial distribution of phase singularities in ventricular fibrillation. Circulation 2003; 108 (3): 354-359. [ Links ]
90. Zaitsev AV, Guha PK, Sarmast F, Kolli A, Berenfeld O, Pertsov AM, et al. Wavebreak formation during ventricular fibrillation in the isolated, regionally ischemic pig heart. Circ Res 2003; 92 (5): 546-553. [ Links ]
91. Liu YB, Pak HN, Lamp ST, Okuyama Y, Hayashi H, Wu TJ, et al. Coexistence of two types of ventricular fibrillation during acute regional ischemia in rabbit ventricle. J Cardiovasc Electrophysiol 2004; 15 (12): 1433-1440. [ Links ]


{kind=link}