










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Colombiana de Cardiología
versión impresa ISSN 0120-5633
Rev. Col. Cardiol. v.15 n.1 Bogota ene./feb. 2008
Reporte de un caso y revisión de la literatura
Report of one case and literature review
(1) Universidad CES, Medellín, Colombia.
(2) Unidad de cuidado intensivo cardiovascular pediátrico, Clínica Cardiovascular Santa María. Medellín, Colombia.
(3) Cardiología pediátrica. Clínica Cardiovascular Santa María. Medellín, Colombia.
Correspondencia: Dr. Alejandro Díaz Díaz. Carrera 63 A No. 32 E 94. Medellín (Antioquia), Colombia. Teléfonos: 2353385 - 2657391 - 3113891754. Correo electrónico: alejodiaz81@hotmail.com.
Recibido: 21/06/2007. Aceptado: 06/03/2008.
El bloqueo aurículo-ventricular completo congénito, es una entidad poco común, que presenta alta morbilidad y mortalidad con incidencia real que permanece desconocida y requiere alto índice de sospecha para su diagnóstico y, por ende, su temprana intervención. Se observa en hijos de madres con enfermedades autoinmunes del tejido conectivo, en especial, lupus eritematoso sistémico cuando su aparición es congénita. A nivel postnatal, es más frecuente que ocurra por cardiopatías congénitas. También puede manifestarse en corazones normales desde el punto de vista estructural. El hallazgo clínico característico es bradicardia persistente que se manifiesta desde la vida intrauterina y repercute en la estabilidad circulatoria del feto llegando a producir hidrops, complicación seria y letal. Después del nacimiento aparece igualmente con bradicardia que puede o no descompensar la parte hemodinámica del paciente. El diagnóstico se hace por sospecha clínica, con ecocardiografía fetal y postnatal, electrocardiograma y detección de anticuerpos maternos tipo antiRo y antiLa. La implantación de un marcapasos es el tratamiento definitivo que contribuye a mejorar la sobrevida y el pronóstico de estos pacientes.
Se presenta el caso de una paciente prematura, de 31 semanas, debido a hidrops no inmune, en quien se diagnosticó bloqueo aurículo-ventricular completo congénito secundario a lupus materno confirmado por anticuerpos anti-nucleares francamente positivos y anticuerpos antiRo y antiLa positivos, quien recibió manejo con soporte inotrópico y posterior implantación de marcapasos. Presentó mejoría completa de la falla cardiaca y se remitió hacia otra institución para manejo convencional del prematuro.
Palabras clave: bloqueo aurículo-ventricular congénito completo, síndrome lúpico neonatal, pretérmino, marcapaso.
Complete congenital atrioventricular block is a rare entity that has a high morbidity and mortality. Its real incidence remains unknown and a high suspicion index is needed for its diagnosis and consequently for its early intervention. It is observed in children of mothers having connective tissue autoimmune diseases, in particular systemic lupus erythematosus, when the condition is congenital. If it is post-natal, congenital cardiopathies are responsible in most cases. It may also appear in structurally normal hearts. The characteristic clinical finding is persistent bradycardia manifested since intrauterine life and affecting the circulatory fetal stability, going as far as to produce hydrops fetalis, a serious and lethal condition. After birth, it appears with bradycardia as well, that may or not unbalance the patient hemodynamics. Diagnosis is made upon clinical suspicion with fetal echocardiography and when post-natal, through electrocardiogram and maternal antibody type antiRo and antiLa. Pacemaker implantation is the definitive treatment that contributes to improve patient survival and prognosis.
We present the case of a premature female patient with 31 weeks of gestation due to non-immune hydrops in who complete atrioventricular block secondary to maternal lupus erythematosus confirmed by frankly positive anti-nuclear antibodies and positive antiRo and antiLa antibodies was diagnosed, and that received inotropic support after pace maker implantation. She improved completely from her heart failure and was sent to other institution for conventional management of prematures.
Key words: congenital complete atrioventricular block, neonatal lupus syndrome, preterm, pace maker.
Reporte de un caso clínico
Paciente recién nacida, producto de un embarazo de 31 semanas, madre de 36 años, hemo-clasificación A (-), Coombs indirecto negativo, VDRL no reactivo, VIH negativo. Embarazo complicado por feto con falla cardiaca manifestada por bradicardia, hidrops no inmune (derrame pleural y pericárdico) por lo cual se realizó cesárea. Apgar no conocido al ingreso en vista de que nació en otra institución, peso: 1.780 gramos, talla: 41 centímetros. Grupo: O Rh positivo.
Desde el nacimiento presentó dificultad respiratoria severa, por lo cual requirió ventilación mecánica con parámetros ventilatorios altos (PIP 30, PEEP 5, ciclaje 60, TI 0,34, FiO2 100%), soporte inotrópico alto (dopamina 10 mcg/kg/min, dobutamina 20 mcg/kg/min) y bradicardia sostenida de 40 latidos por minuto. Se diagnosticó bloqueo aurículo-ventricular completo congénito. Por tal motivo, en la madre se iniciaron estudios por sospecha de lupus eritematoso sistémico el cual se confirmó por anticuerpos antinucleares (ANAS) positivo, patrón moteado, 1:320, antiRo 141u/lL (Valor normal 0-15), antiLa 124 u/L (VN 0-15), anticardiolipinas IgM 2,5 u/L (VN 0-7) e IgG 586 u/L (VN 0-10). Con estos exámenes se confirmó el diagnóstico de lupus eritematoso sistémico materno y el de síndrome lúpico neonatal como causa del bloqueo.
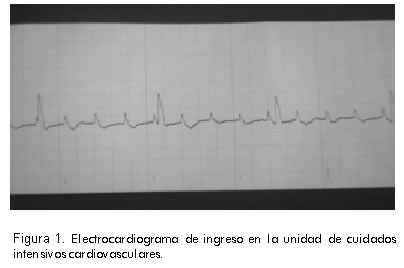
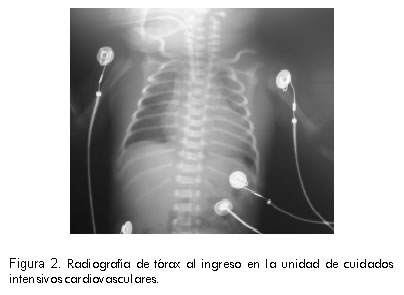
Por persistir con bradicardia severa e inestabilidad hemodinámica, en su segundo día de vida la paciente se remitió a la Clínica Cardiovascular. Llegó en malas condiciones, mal perfundida, ictérica, con acidosis metabólica severa. El electrocardiograma del ingreso confirmó el diagnóstico de bloqueo aurículo-ventricular completo congénito con frecuencia auricular de 170 por minuto y frecuencia ventricular de 45 por minuto (Figura 1). La radiografía de tórax al ingreso demostró cardiomegalia y signos de edema pulmonar secundario a la disfunción ventricular (Figura 2).
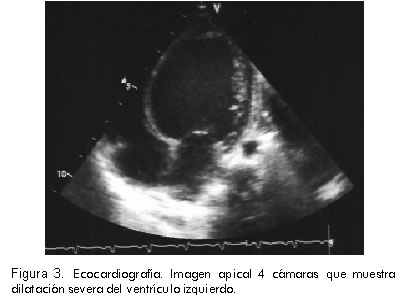
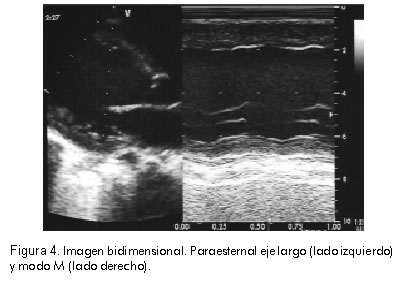
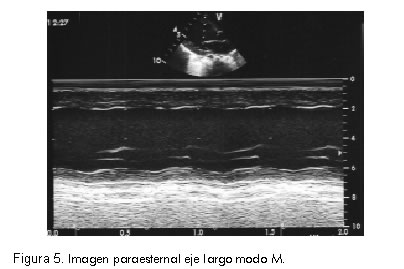
Se tomó ecocardiografía que informó dilatación biventricular, disfunción ventricular izquierda con fracción de eyección de 27% y disfunción diastólica del ventrículo izquierdo. Movimiento paradójico del septum interventricular. Insuficiencia tricúspide moderada con gradiente máxima de 40 mm Hg que permitía calcular una presión de la arteria pulmonar de 50 mm Hg. Foramen ovale permeable con cortocircuito bidireccional. Conducto arterioso permeable de 2,8 mm con cortocircuito de izquierda a derecha (Figuras 3, 4 y 5). Se inició manejo con dopamina, dobutamina, isoproterenol, furosemida y calcio. Además, la paciente presentaba disfunción renal por aumento de las cifras de creatinina y poliuria.
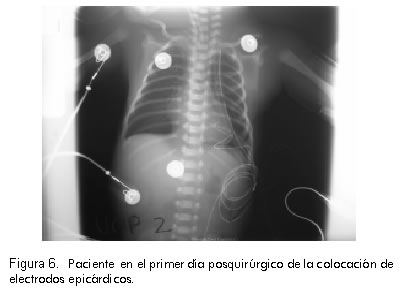
Se le colocaron electrodos epicárdicos para manejo con marcapasos externo transitorio urgente hasta su estabilización hemodinámica. Luego del procedimiento, la paciente mostró mejoría notoria de su condición clínica, lo cual permitió desmontar el soporte inotrópico y efectuar la extubación (Figura 6).
Se tomaron anticuerpos antiRo y antila, los cuales fueron positivos; 126 y 104 unidades, respectivamente.
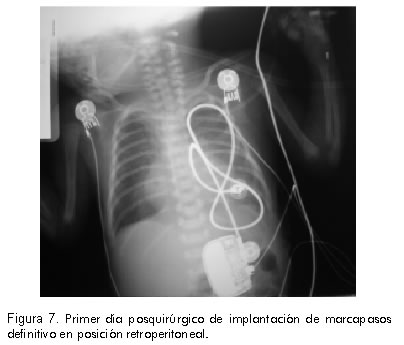
En su noveno día de vida, se le implantó un marcapasos definitivo con electrodo pericárdico y la unidad en retroperitoneo, sin presentar complicaciones operatorias (Figura 7).
La ecocardiografía de control reportó insuficiencia tricúspide trivial, estenosis ligera de rama izquierda de arteria pulmonar, FOP con shunt bidireccional de predominio izquierda a derecha, dilatación leve del ventrículo izquierdo, fracción de eyección de 36% y disfunción sisto-diastólica del ventrículo izquierdo. Movimiento paradójico del septum interventricular.
Egresó de la institución en el décimo tercer día de vida y se remitió a otra entidad hospitalaria para continuar con el manejo de recién nacido prematuro. Se prescribió con enalapril, oxígeno y medicamentos para bebé prematuro. En el momento en aumento de vía oral y con nutrición parenteral. En la figura 8 se puede observar a la paciente a su egreso de la institución.

Bloqueo aurículo-ventricular congénito completo
En 1901, Morquio describió y reconoció esta enfermedad completo congénito y además describió la ocurrencia familiar y su relación con la crisis de Stokes-Adams y la muerte neonatal (1).
Es un desorden poco común con incidencia de aproximadamente 1/15.000 - 1/22.000 nacidos vivos (2). Sin embargo y a pesar de los medios actuales para el diagnóstico prenatal temprano (3), aún es una patología con alta morbilidad y mortalidad con incidencia real que permanece desconocida y que requiere alto índice de sospecha para su diagnóstico e intervención temprana.
Etiología
El síndrome lúpico neonatal produce 70% a 95% de los casos de bloqueo aurículo-ventricular completo congénito que se manifiestan in útero o en el período neonatal inmediato, pero sólo 5% de los casos postnatales tardíos (4,5), en donde las cardiopatías congénitas, específicamente la transposición «corregida» de grandes arterias y los defectos septales atrioventriculares, son la causa de más del 50% de los casos (6,7).
El síndrome lúpico neonatal es una enfermedad autoinmune que se produce por el paso de anticuerpos maternos del tipo antiRo/SSA y antiLa/SSB (anticuerpos contra antígenos extraíbles del núcleo, ENA), cuya principal complicación y una de las manifestaciones que definen el cuadro, es el bloqueo aurículo-ventricular completo congénito (8). Aunque la entidad se describió inicialmente en hijos de madres con lupus eritematoso sistémico (9), ésta puede producirse en el contexto de otras enfermedades del tejido conectivo que produzcan este tipo de autoanticuerpos, como por ejemplo el síndrome de Sjögren (10). A pesar de que toda madre de un niño con síndrome lúpico neonatal tiene anticuerpos circulantes, sólo 1% a 2% de madres con anticuerpos, tendrá un hijo con bloqueo aurículo-ventricular completo congénito (11).
En 10% de los casos, la enfermedad se presentará en un corazón con estructura normal y sin asociaciones a enfermedades del tejido conectivo o anticuerpos circulantes en la madre.
Patogénesis
Aún no se entiende con exactitud el mecanismo exacto por el cual se produce el bloqueo; sin embargo, el paso de autoanticuerpos a través de la placenta de madres con enfermedad autoinmune, fenómeno que empieza a manifestarse desde la semana 12 de gestación, se acepta como el más probable (1, 12). Los anticuerpos antiRo y antiLa actúan contra las proteínas ribonucleares «Ro» y «La» intracelulares, importantes para la maduración de RNA polimerasas. Estos autoanticuerpos tienen especial afinidad por células del sistema de conducción fetal, sobre todo entre las semanas 16 y 24 de gestación (13,14). Allí desencadenan un proceso autoinmune que lesiona el nodo aurículo-ventricular y los tejidos circundantes causando isquemia, necrosis y posterior fibrosis; además, se crea una desconexión entre la aurícula y el nodo aurículo-ventricular (15). Tampoco es muy claro cómo se desencadena este proceso inflamatorio. Se cree que la apoptosis del miocito favorece la expresión de estas proteínas nucleares para que las reconozcan los autoanticuerpos, generando inducción de la inflamación, fagocitosis de la célula apoptótica y liberación de factor de necrosis tumoral (16). Otra teoría, es el posible efecto arritmogénico de los autoanticuerpos al inhibir, de forma directa, los canales de calcio tipo L, los cuales son fundamentales para el inicio y la propagación del potencial de acción en el nodo aurículo-ventricular (17).
Clínica
Las manifestaciones dependen de la edad de presentación. Puede aparecer in útero, al nacimiento o más tarde (8). Cuando se presenta in útero, casi invariablemente es por un síndrome lúpico neonatal. La bradicardia fetal puede detectarse desde la semana 18 hasta la 28, por medio de ecocardiografía fetal con la cual puede medirse el intervalo PR mecánico (18). Los fetos con frecuencias más bajas tienen grandes complicaciones como aborto espontáneo, hidrops severo, fibroelastosis endocárdica, derrame pericárdico y muerte intrauterina (1, 4). Si logran alcanzar el nacimiento, tienen mortalidad de hasta 30% en un neonato mayor de 34 semanas y hasta de 50% en menores (19). Estos niños desarrollarán cardiomiopatía dilatada, fibroelastosis e insuficiencia mitral severas.
Cuando se manifiesta en el período neonatal, la bradicardia es el signo cardinal, además de los signos de falla cardiaca secundarios a bajo gasto.
El electrocardiograma mostrará frecuencia atrial aumentada, frecuencia ventricular baja y ritmo de la unión o del nodo aurículo-ventricular de escape o ectópico. Puede encontrarse bloqueo aurículo-ventricular de primer o segundo grado que posteriormente se convertirá en completo. Hasta 40% de los bloqueos aurículo-ventriculares completos congénitos pueden no manifestarse hasta la niñez. Esto tal vez se debe a que pueden presentarse de forma intermitente y asintomática. En esta situación es muy poco probable que la causa sea el síndrome lúpico neonatal (20).
Diagnóstico
Aunque la historia prenatal será de gran ayuda para el diagnóstico, en más de 50% de los casos la madre no tiene enfermedad activa, sólo anticuerpos circulantes, y únicamente un tiempo después desarrollarán la enfermedad (21). La mayor concentración de autoanticuerpos en el sistema de conducción fetal, se da entre las semanas 16 y 24 de gestación. Este es el período de mayor vulnerabilidad para el feto. Aunque no hay guías establecidas, en las embarazadas en riesgo se recomienda hacer seguimiento con ecocardiografía fetal cada semana durante este período y luego cada dos semanas hasta la 32; después de ésta es poco probable que se produzca bloqueo (22).
Un feto en el que se detecte bloqueo aurículo-ventricular completo congénito, deberá nacer en un centro de alta complejidad donde pueda recibir atención en un servicio de cardiología pediátrica. En el neonato, bastará tener una frecuencia cardiaca baja no explicada por otra causa, para sospechar el diagnóstico. Es obligatorio realizar electrocardiograma después del nacimiento para corroborar el bloqueo. Debido a que más del 90% de los casos ocurren por síndrome lúpico neonatal, también es imperativo efectuar estudios a la madre en caso de que no tenga enfermedad manifiesta ni anticuerpos medidos, ya que es el incremento de los autoanticuerpos séricos en ella el que hará el diagnóstico de síndrome lúpico neonatal. No tiene utilidad medir anticuerpos en el neonato (1-4).
Tratamiento prenatal
El objetivo de realizar ecocardiografías seriadas en la madre, es detectar alteraciones fetales que podrían evolucionar a bloqueo aurículo-ventricular completo congénito de forma temprana con el fin de seleccionar candidatas al tratamiento preventivo. El tratamiento con esteroides fluorinados se fundamenta en el hecho de contrarrestar el proceso inflamatorio generado por los anticuerpos. No obstante, aún es controversial por los efectos secundarios en la madre y por el riesgo de hipoplasia e insuficiencia adrenal en el feto y el neonato. Se describe el uso de dexametasona o betametasona en madres con fetos que presenten bloqueo completo o incompleto con signos de hidrops, iniciándolos lo más pronto posible y hasta el final del embarazo. Si en cinco semanas no hay mejoría, se suspende el tratamiento (23).
Cuando el bloqueo es completo, no tiene utilidad dar tratamiento prenatal ya que la tasa de mejoría es casi nula. No se indica el manejo preventivo en madres sólo con anticuerpos positivos sin alteraciones en el feto, por el bajo riesgo de desarrollar el cuadro y por los efectos secundarios (24).
Tratamiento posnatal
En niños que inicialmente sean asintomáticos, se vigila, durante la primera semana, la aparición de síntomas o la progresión de un bloqueo aurículo-ventricular de primer o segundo grado hacia uno de tercero. El tratamiento consiste en el manejo de la falla cardiaca por medio de medicamentos y soporte inotrópico. En más del 60% de los pacientes se implantará marcapasos definitivo; más de la tercera parte lo requieren en la primera semana de vida (1).
Las recomendaciones para el empleo de marcapasos figuran dentro de las guías del Colegio Americano de Cardiología (ACC), la Asociación Americana del Corazón (AHA) y la Sociedad Norteamericana de Electrofisiología y Marcapasos para la implantación de marcapasos (25). Las indicaciones de clase I (evidencia demostrada o acuerdo general para el uso indiscutible) en niños con bloqueo aurículo-ventricular II avanzado y de III grado, son:
- Bradicardia sintomática, disfunción ventricular o bajo gasto.
- Ritmo de escape con QRS amplio, taquicardia ectópica de la unión, arritmia ventricular compleja.
- Frecuencia cardiaca menor de 50/minuto en reposo o menor de 70/minuto si hay cardiopatía congénita.
- QT prolongado.
- Taquicardia ventricular sostenida con o sin QT prolongado.
- Bloqueo aurículo-ventricular completo después de siete días posquirúrgico.
Pronóstico
Además de las complicaciones intrauterinas ya mencionadas, existe riesgo de desarrollar cardiomiopatía dilatada en 23% de los pacientes a pesar de la implantación temprana de marcapasos. Los pacientes que permanecen asintomáticos en la niñez, pueden desarrollar limitación al ejercicio con el paso de los años y 5% a 11% desarrollarán falla cardiaca a largo plazo.
La mortalidad oscila entre 20% a 30% si no se instaura un manejo oportuno.
Los principales factores asociados con mal pronóstico son: desarrollo de bloqueo in útero, frecuencias menores de 55/minuto, presencia de hidrops, bajo peso al nacer, prematurez, género masculino y desarrollo de cardiomiopatía dilatada en quienes la mortalidad será mayor a 60% (4, 26).
El seguimiento se hace mínimo cada año con electrocardiograma, monitoreo Holter y ecocardiografía de control, sobre todo en pacientes sin marcapasos.
Discusión y conclusiones
El caso en mención corresponde claramente al de un síndrome lúpico neonatal de acuerdo con los parámetros de diagnóstico clínico, radiológico, electrocardiográfico y ecocardiográfico. Las indicaciones para la implantación de marcapasos definitivo se difunden y reportan ampliamente en la literatura, ya que son la única medida que ha logrado cambiar la sobrevida de los pacientes (1). Desde el punto de vista técnico, tiempo atrás era difícil realizar este procedimiento en niños pues los dispositivos con los que se contaba eran diseñados para adultos y los de menor tamaño sólo podían emplearse en niños pre-escolares, pues en neonatos, especialmente en los prematuros, era complicado encontrar un lugar anatómico en el cual colocar la unidad de marcapasos, habitualmente en la pared abdominal anterior, posterior a los rectos anteriores. Pese a ello el riesgo de complicaciones como necrosis de la piel, extrusión de la unidad, entre otras, era frecuente. En la actualidad, con los avances en la tecnología y con la aparición de dispositivos cada vez más pequeños, es posible brindar este tratamiento a ese tipo de pacientes. En nuestro medio no se encuentran datos al respecto.
En cuanto a la literatura mundial, existen diversos reportes de implantación de marcapasos definitivo en recién nacidos a término y prematuros a nivel abdominal y retroperitoneal, con tasas de éxito y sobrevida muy aceptables (27-28). Existen reportes de neonatos menores de 1.500 gramos (30, 31, 32), incluso de 30 semanas y 759 gramos, con resultados satisfactorios a un año de seguimiento (33). Se espera que con la detección de más casos de una patología no muy frecuente, más la experiencia y la llegada de dispositivos de tamaño óptimo, pueda brindársele una excelente atención a estos pacientes y así velar por su mejor pronóstico y sobrevida a largo plazo.
Puede concluirse que:
- El bloqueo aurículo-ventricular completo congénito es una entidad neonatal poco frecuente.
- Debe sospecharse en toda embarazada con enfermedad autoinmune manifiesta o con antiRo y antiLa positivos.
- Puede sospecharse desde la vida in útero por ecocardiografía fetal.
- El manejo definitivo de los pacientes con esta entidad, es la implantación de marcapaso definitivo, siempre y cuando tenga indicación clínica. Esto es lo único que ha logrado mejorar el pronóstico y la sobrevida de los pacientes.
- Hoy en día la edad gestacional y el peso, no son contraindicaciones para realizar este procedimiento.
- El paciente que se reporta en nuestra revisión tiene un diagnóstico confirmado de síndrome lúpico neonatal por historia clínica, laboratorio, ecocardiografía y electrocardiografía. Recibió manejo de acuerdo con lo estandarizado en la literatura mundial y se obtuvo un resultado exitoso.
Bibliografía
1. Jayaprasad M, Johnson F, Venugopal M. Congenital complete heart block and maternal connective tissue disease. Internal J Cardiol 2006; 112: 153-8. [ Links ]
2. Michaelsson M, Engle MA. Congenital complete heart block: an international study of the natural history. Cardiovasc Clin 1972; 4: 85-101. [ Links ]
3. Sonesson SE, Salomonsson S, Jacobsson LA, et al. Signs of first-degree heart block occur in one-third of fetuses of pregnant women with anti-SSA/Ro 52-kd antibodies. Arthritis Rheum 2004; 50: 1253-61. [ Links ]
4. Jaeggi ET, Hamilton RM, Silverman ED, et al. Outcome of children with fetal, neonatal or childhood diagnosis of isolated congenital atrioventricular block. J Am Coll Cardiol 2002; 39: 130. [ Links ]
5. Johansen AS, Herlin T. Neonatal lupus syndrome. Association with complete congenital atrioventricular block. Ugeskr Laeger 1998; 160 (17): 2521-5. [ Links ]
6. Jaeggi ET, Hornberger LK, Smallhorn JF, Fouron JC. Prenatal diagnosis of complete atrioventricular block associated with structural heart disease: combined experience of two tertiary care centers and review of the literature. Ultrasound Obstet Gynecol 2005; 26: 16-21. [ Links ]
7. Berg C, Geipel A, Kohl T, et al. Atrioventricular block detected in fetal life: associated anomalies and potential prognostic markers. Ultrasound Obstet Gynecol 2005; 26: 4 -15. [ Links ]
8. Kertesz N, Fenrich AL, Friedman RA. Congenital complete atrioventricular block. Tex Heart Inst J 1997; 24: 301-7. [ Links ]
9. Hull D, Binns BAO, Joyce D. Congenital heart block and widespread fibrosis due to maternal erythematosus. Arch Dis Child 1966; 41: 688-90. [ Links ]
10. McCue CM, Mantakas ME, Tingelstad JB, Ruddy S. Congenital heart block in newborns of mothers with connective tissue disease. Circulation 1977; 56: 82-90. [ Links ]
11. Brucato A, Frassi M, Franceschin F, et al. Risk of congenital complete heart block in newborns of mothers with anti-Ro/SSA antibodies detected by counterimmunoelectrophoresis: a prospective study of 100 women. Arthritis Rheum 2001; 44: 1832. [ Links ]
12. Buyon JP, Winchester RJ. Complete congenital block: a model of passively acquired autoimmunity. Arthritis Rheum 1990; 33: 609-14. [ Links ]
13. Alexander E, Buyon JP, Provost TT, et al. Anti-Ro/SS-A antibodies in the pathophysiology of congenital heart block in neonatal lupus syndrome: an experimental model. Arthritis Rheum 1992; 35: 176-189. [ Links ]
14. Buyon JP, Kim MY, Copel JA, Friedman DM. Anti-Ro/SSA antibodies and congenital heart block: necessary but not sufficient. Arthritis Rheum 2001; 44 (8): 1832-5. [ Links ]
15. Ho SY, Esscher E, Anderson RH, Michaelsson M. Anatomy of congenital complete heart block and relation to maternal anti-Ro antibodies. Am J Cardiol 1986; 58: 291-4. [ Links ]
16. Miranda-Carus ME, Askanase AD, Clancy RM, et al. Anti-SSA/Ro and anti-SSB/La auto antibodies bind the surface of apoptotic fetal cardiocytes and promote secretion of TNF-alpha by macrophages. J Immunol 2000; 165: 5345-51. [ Links ]
17. Xiao GQ, Hu K, Boutjdir M. Direct inhibition of expressed cardiac L- and T-type calcium channels by IgG from mothers whose children have congenital heart block. Circulation 2001; 103: 1599-1604. [ Links ]
18. Rosenthal D, Friedman DM, Buyon J, Dubin A. Validation of the Doppler PR interval in the fetus. J Am Soc Echocardiogr 2002;15: 1029-30. [ Links ]
19. Buyon JP, Hiebert R, Copel J, et al. Autoimmune-associated congenital heart block: demographics, mortality, morbidity and recurrence rates obtained from a National Neonatal Lupus Registry. J Am Coll Cardiol 1998; 31: 1658. [ Links ]
20. Michaelsson M, Jonzon A, Riesenfeld T. Isolated congenital complete atrioventricular block in adult life. A prospective study. Circulation 1995; 92: 442. [ Links ]
21. Waltuck J, Buyon JP. Autoantibody-associated congenital heart block: outcome in mothers and children. Ann Intern Med 1994; 120: 544-51. [ Links ]
22. Brucato A, Cimaz R, Stramba-Badiale M. Neonatal lupus. Clin Rev Allergy Immunol 2002; 3: 279-99. [ Links ]
23. Breur JMPJ, Visser GHA, Kruize AA, Stoutenbeek P, Meijboom EJ. Treatment of fetal heart block with maternal steroid therapy: case report and review of the literature. Ultrasound Obstet Gynecol 2004; 24: 467-72. [ Links ]
24. Ho SY, Esscher E, Anderson RH, Michaelsson M. Anatomy of congenital complete heart block and relation to maternal anti-Ro Costedoat-Chalumeau N, Amoura Z, Le Thi Hong D, et al. Questions about dexamethasone use for the prevention of anti-SSA related congenital heart block. Ann Rheum Dis 2003; 63: 1010-2. [ Links ]
25. Gregoratos G, Abrams J, Epstein AE, et al. ACC/AHA/NASPE 2002 Guideline Update for Implantation of Cardiac Pacemakers and Antiarrhythmia Devices-summary article: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2002; 40 (9): 1703-19. [ Links ]
26. Groves AMM, Allan LD, Rosenthal E. Outcome of isolated congenital complete heart block diagnosed in utero. Heart 1996; 75: 190-4. [ Links ]
27. Tomita Y, Imoto Y, Tominaga R, Yasui H. Successful implantation of a bipolar epicardial lead and autocapture pacemaker in a low body weight infant with congenital atrioventricular block. Report of a case. Surg Today 2000; 30: 555- 557. [ Links ]
28. Rodríguez S, Villegas O, Martínez R. Placement of permanent epicardial pacemaker in a newborn with congenital complete AV block. Arch Inst Cardiol Mex 2000; 70 (2): 180-6. [ Links ]
29. Sosa E, Van Doesburg N, Kratz C, Stanley P, Chartrand C. Implantation of a permanent pacemaker in a premature Infant. Tex Heart Inst J 1988; 15: 128-130. [ Links ]
30. Khositseth A, Samankatiwat P, Withurawanit W, Khowsathit P. Pacing in preterm with hydrops fetalis due to congenital complete heart block. Asian Cardiovasc Thorac Ann. 2006; 14 (5): 428-31. [ Links ]
31. Takabayashi S, Ito H, Shimpo H, Sawada H, Mitani Y, Komada Y. Emergent permanent pacemaker implantation in a premature 1,502 g neonate. Jpn J Thorac Cardiovasc Surg. 2005; 53 (4): 199-201. [ Links ]
32. von Schnakenburg C, Fink C, Peuster M, Wessel A, Vázquez-Jiménez J. Permanent pacemaker implantation in a 1,445 g preterm neonate on the first day of life. Thorac Cardiovasc Surg 2002; 50 (6): 63-5. [ Links ]
33. Quek SC, Low KT, Sim EK, Joseph R. A case report on the perinatal management of a 30-week preterm baby with congenital complete heart block. Ann Acad Med Singapore 2000; 29 (4): 510-3. [ Links ]

