










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Cardiología
Print version ISSN 0120-5633
Rev. Col. Cardiol. vol.15 no.4 Bogota Jul./Aug. 2008
Departamento de Hemodinamia, Fundación Clínica Abood Shaio, Bogotá, DC., Colombia.
Correspondencia: Departamento de Hemodinamia, Fundación Clínica Abood Shaio. Diagonal 115 A No.70C - 75, Bogotá, DC. Colombia.. Teléfono: 2714323.
Recibido: 16/05/07. Aprobado: 27/05/08.
El ácido acetil salicílico es quizás uno de los medicamentos más antiguos conocidos en el mundo, desde los tiempos de Hipócrates cuando se consumía la corteza del Sauce por sus efectos analgésicos y antipiréticos. Sus propiedades antiplaquetarias fueron reconocidas por primera vez en 1967, y su mecanismo de acción fue dilucidado en 1971. Desde entonces, se usa más por esas propiedades antiplaquetarias que por su efecto analgésico o antipirético.
La activación y agregación plaquetaria desempeña un rol importante en la patogénesis de la trombosis arterial, lo cual conduce a síndrome coronario agudo y a complicaciones trombóticas durante y después de intervenciones coronarias percutáneas, de ahí que el ácido acetil salicílico sea el agente antiplaquetario más empleado.
Estudios clínicos demuestran su eficacia tanto en prevención primaria como en secundaria de infarto del miocardio, accidente cerebrovascular y muerte cardiovascular. A pesar de sus probados beneficios, el riesgo relativo de eventos vasculares recurrentes entre los pacientes que lo toman permanece relativamente alto, y se estima en 8% a 18% después de dos años. La resistencia terapéutica al ácido acetil salicílico podría explicar en parte este riesgo.
Aunque aún no se establecen los criterios de diagnóstico formal y un sistema válido de medición, la resistencia al ácido acetil salicílico puede afectar entre 5% a 45% de la población. Dada la prevalencia de la enfermedad cardiovascular, el impacto potencial de la resistencia a este medicamento es amplio.
Otra clase de agentes que actúan mediante el bloqueo de la agregación plaquetaria son los derivados tienopiridínicos, que incluyen el clopidogrel.
El ácido acetil salicílico y el clopidogrel, se convirtieron en la terapia de elección en pacientes que serán sometidos a intervención coronaria con stent. Sin embargo, existe una considerable heterogeneidad a la respuesta individual de los pacientes expuestos a estos fármacos.
Datos actuales muestran que cerca de 4% a 30% de los pacientes tratados con dosis convencionales de clopidogrel, no tienen una adecuada respuesta antiplaquetaria. La resistencia al clopidogrel es un término muy usado que aún precisa de una definición clara. Aun así, se emplea para reflejar que el clopidogrel falla en alcanzar su efecto antiagregante.
Esta revisión discute la evidencia actual con relación a la variabilidad de la respuesta antiplaquetaria de estos dos medicamentos.
Palabras clave: resistencia, ácido acetil salicílico, clopidogrel, antiplaquetarios, plaquetas.
Acetylsalicylic acid is perhaps one of the most antique drugs known worldwide since the time of Hippocrates, when willow bark was used for its analgesic and antipyretic effects. Its antiplatelet properties were known for the first time in 1967 and its mechanism of action was explained in 1971. Since then it is used mostly for its antiplatelet properties than for its analgesic or antipyretic effect.
Platelet activation and aggregation play an important role in arterial thrombosis pathogenesis, which leads to acute coronary syndrome and thrombotic complications during and after percutaneous coronary interventions; for this reason, acetylsalicylic acid is the most used antiplatelet agent.
Clinical trials have shown its efficacy both in primary and secondary prevention of myocardial infarction, stroke and cardiovascular death. Despite its proven benefits, the relative risk of recurrent vascular events among patients taking it remains relatively high and is estimated in 8% to 18% after two years. Therapeutic resistance to acetylsalicylic acid could partially explain this risk.
Even though formal diagnostic criteria and a valid measurement system are not yet established, resistance to acetylsalicylic acid may affect 5% to 45% of the population. Given the prevalence of cardiovascular disease, the potential impact of resistance to this drug is wide.
Another kind of agents that act blocking platelet aggregation are the thienopyridine derivates, that include clopidogrel.
Acetylsalicylic acid and clopidogrel became the election therapy in patients that will undergo coronary intervention with stent implantation. However, there is a considerable heterogeneity to the individual response of patients exposed to these drugs.
Actual data show that almost 30% to 40% of patients treated with conventional doses of clopidogrel do not have an adequate antiplatelet response. Resistance to clopidogrel is a fairly used term that still needs a clear definition. Even so, it is used to reflect that clopidogrel fails in achieving its antiaggregation effect.
This review discusses current evidence in relation to the variability of the antiplatelet response of these two drugs.
Key words: resistance, acetylsalicylic acid, clopidogrel, antiplatelets, platelets.
Introducción
La mayoría de los síndromes coronarios agudos y otras manifestaciones de la enfermedad aterosclerótica, son causados por ruptura o fisura de una placa que produce una consecuente formación de un trombo suboclusivo u oclusivo, en donde la agregación plaquetaria ejerce un papel muy importante (1, 2). La terapia antiplaquetaria es la piedra angular en el tratamiento de las enfermedades cardiovasculares. El ácido acetil salicílico y el clopidogrel emergieron como excelente opción terapéutica en la prevención y el tratamiento de enfermedades del sistema cardiovascular. A pesar de su eficacia, los pacientes que ingieren estos medicamentos, presentan complicaciones y alta morbilidad cardiovascular (3-5). En la actualidad, millones de pacientes reciben bajas dosis de antiagregantes plaquetarios, pero no se sabe cuantos están en régimen de adecuada dosis o medicación equivocada (6).
La resistencia al ácido acetil salicílico y al clopidogrel surge como entidad clínica con consecuencias potencialmente severas, como infarto del miocardio recurrente, accidente cerebrovascular y muerte (6). El mecanismo de resistencia a estos medicamentos es indefinido, pero existen factores clínicos, celulares y genéticos que influyen en la falla terapéutica (7-11). Si se entienden los mecanismos relacionados con la falla terapéutica y si se mejoran los métodos diagnósticos, puede emerger una nueva era de terapia antiplaquetaria individualizada, con mediciones rutinarias de la actividad plaquetaria en la misma forma que las mediciones de colesterol, tensión arterial o glucosa en sangre, con el objetivo de mejorar el cuidado de millones de pacientes.
Definición
En su sentido más amplio, la resistencia clínica al ácido acetil salicílico se refiere a pacientes que presentan eventos isquémicos a pesar de recibirlo. Desde el punto de vista del laboratorio, se refiere a quienes no logran un adecuado grado de inhibición plaquetaria con el medicamento (7). Otros autores la definen como la imposibilidad del ácido acetil salicílico en reducir la producción de tromboxano A2 y, por ende, la agregación y activación plaquetaria (8) (Figura 1).
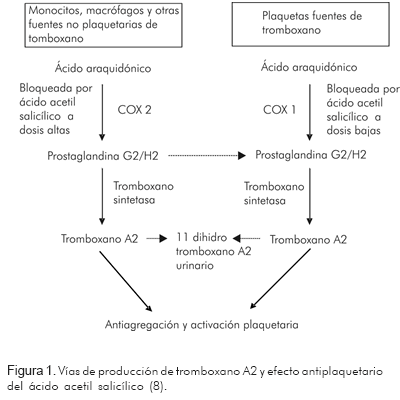
Las mediciones de agregación plaquetaria, activación plaquetaria y tiempo de sangría confirman la variabilidad de la respuesta antitrombótica a la terapia con ácido acetil salicílico (3, 11).
Adhesión plaquetaria, activación y agregación
Cuando se altera la íntima de un vaso, como ocurre en la ruptura de una placa aterosclerótica, se exponen el colágeno subendotelial y el factor de von Willebrand (vWF) a la sangre circulante. Las plaquetas en la sangre se adhieren al colágeno subendotelial y al factor de vWF a través de su receptor de glicoproteína Ia/IIa y Ib/V/IX (12).
La adhesión plaquetaria estimula la activación plaquetaria, la cual conlleva un cambio en su estructura y en la liberación de calcio dentro de la plaqueta. El incremento de la concentración del ión calcio dentro de la plaqueta, tiene varias consecuencias (8):
1. Induce a un cambio conformacional de la plaqueta exteriorizando sus receptores IIb/IIIa en su superficie, para así unirse a proteínas como el fibrinógeno, en la circulación.
2. Cataliza la liberación de moléculas activas entre ellas el adenosindifosfato (ADP) de los gránulos plaquetarios a la circulación, donde pueden unirse a sus receptores en otras plaquetas y desencadenar su activación.
3. Promueve la acción de la fosfolipasa A2 para producir ácido araquidónico.
El ácido araquidónico en las plaquetas es convertido en tromboxano A2 (TXA2) en una reacción que es catalizada por la enzima ciclo-oxigenasa 1 (COX1) (para formar prostaglandina G2/H2) y por la tromboxano sintetasa para formar TXA2 (Figura 1). El TXA2 incrementa la expresión de los receptores de fibrinógeno en la membrana plaquetaria y además es liberado a la circulación uniéndose a sus receptores en las plaquetas adyacentes para desencadenar su activación; posteriormente, el TXA2 es convertido en tromboxano B2 (TXB2), el cual puede ser medido en sangre (8).
Farmacocinética del ácido acetil salicílico
El ácido acetil salicílico es rápidamente absorbido en el estómago y el duodeno, por difusión pasiva como ácido acetil salicílico no disociado a través de la membrana gastrointestinal. Cuando se ingiere sin cubierta entérica alcanza su pico plasmático en 30 a 40 minutos, en contraste con las tres a cuatro horas después de la ingestión de la presentación con cubierta entérica (15), así que los pacientes deben masticar estas presentaciones para alcanzar un rápido efecto antiplaquetario.
El ácido acetil salicílico entra en contacto por primera vez con las plaquetas en la circulación portal; su vida media es de 15 a 20 minutos ya que es rápidamente hidrolizada a ácido salicílico (16). Aunque su vida media es corta, la inhibición de la función plaquetaria es evidente al cabo de 40 a 60 minutos y su efecto antiplaquetario persiste durante toda la vida de la plaqueta (7 a 9 días). Teniendo en cuenta que 10% de las plaquetas son reemplazadas cada 24 horas, puede asumirse que aproximadamente en cinco a seis días de interrumpirse la administración de ácido acetil salicílico, 50% de las plaquetas tendrá un funcionamiento normal (16).
Mecanismo de acción
El mecanismo de acción del ácido acetil salicílico consiste en la inhibición irreversible de la enzima ciclo-oxigenasa (COX) de las plaquetas, con lo que se interrumpe la transformación de ácido araquidónico en TXA2, compuesto con una gran actividad vasoconstrictora y un potente efecto de agregación plaquetaria (15) (Figura 1).
El ácido acetil salicílico a dosis bajas (30 a 325 mg) inhibe a la COX 1 plaquetaria condicionando su efecto antiagregante y a dosis más altas (500-1.300 mg/día) inhibe a la ciclo-oxigenasa 2 (COX2) condicionando su efecto analgésico y antipirético (17) (Figura 1).
Aunque el TXA2 es un prostanoide derivado, en su mayoría, de la COX1 plaquetaria, su biosíntesis es muy sensible a la inhibición del ácido acetil salicílico a dosis bajas; por el contrario, la prostaciclina (PGI2), la cual tiene un efecto inverso al del TXA2, es decir, inhibe la agregación plaquetaria e induce la vasodilatación, es derivada predominantemente de la COX2, la cual es inhibida por el ácido acetil salicílico a dosis altas (18). El efecto anti-inflamatorio relativamente débil del ácido acetil salicílico a baja dosis (100-325 mg), se explica, en parte, porque éste ejerce una inhibición 170 veces más potente sobre la COX1 que sobre la COX2 (21).
Cabe resaltar que la administración conjunta de inhibidores reversibles de la COX1, como el ibuprofeno y el naproxeno, pueden prevenir la acetilación irreversible de la COX1 plaquetaria por el ácido acetil salicílico, por un mecanismo de competencia por el residuo de serina en la posición 529 del receptor plaquetario (19, 20). Esta interacción farmacológica no ocurre con otros medicamentos anti-inflamatorios no esteroides, como el diclofenaco, que posee cierto grado de selectividad COX2 (19).
El ácido acetil salicílico acetila de manera irreversible a la COX1 y suprime así la producción de TXA2, el cual es uno de los más potentes agonistas de la agregación plaquetaria, con lo cual se previene la formación de trombos a través de este mecanismo.
Factores implicados en la resistencia al ácido acetil salicílico
Aunque el efecto antiplaquetario del ácido acetil salicílico no es uniforme en todas las plaquetas, la inhibición de la actividad plaquetaria que ejerce, está sujeta a variables que dependen de cada individuo. Esta respuesta impredecible al ácido acetil salicílico puede atribuirse a factores clínicos, celulares y genéticos (7, 8) (Figura 2).
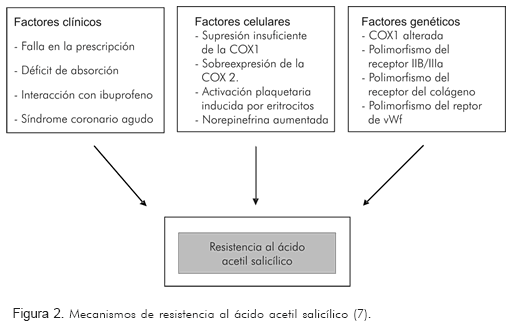
Factores clínicos
Pueden variar desde el incumplimiento del tratamiento por parte del paciente, hasta la falta de prescripción adecuada por parte del médico. Otros pacientes pueden tomarlo pero no tienen una adecuada absorción (22). Se pueden presentar interacciones farmacológicas como con el ibuprofeno, como ya se había anotado. El síndrome coronario agudo y la insuficiencia cardiaca congestiva, se asocian con aumento en la reactividad plaquetaria (23). La hiperglucemia puede disminuir la efectividad de la actividad antiplaquetaria del ácido acetil salicílico al incrementar la cantidad de sustancias oxidantes (6). Así mismo, el aumento de catecolaminas asociado con estrés, puede afectar la respuesta plaquetaria (6).
Factores celulares
En éstos se incluyen la inadecuada supresión de la COX1 plaquetaria; la resistencia al ácido acetil salicílico se atribuye a una sobre-expresión del ARNm de la COX2 por células endoteliales y plaquetas. Las resolvinas, una familia bioactiva de metabolitos de los ácidos omega 3, son mediáticas de la respuesta inflamatoria y se generan por la acetilación de la COX2 por el ácido acetil salicílico. De hecho, la deficiencia de estas sustancias puede conducir a falla en la terapéutica (6).
Factores genéticos
Desempeñan un papel importante en la respuesta terapéutica, como un polimorfismo de las glicoproteínas Pl (A1/A2) de la membrana plaquetaria que se asocian con una respuesta atenuada al ácido acetil salicílico (24). El polimorfismo del factor de von Willebrand o del gen del receptor del colágeno, también se postula como causa de la resistencia al ácido acetil salicílico (25).
Existen otros factores que pueden afectar la respuesta de la terapia antiplaquetaria y de los que los exámenes de laboratorio usados a la cabecera del paciente no pueden detectar su impacto (8); éstos son:
Biodisponibildad reducida
- Ingestión inadecuada de ácido acetil salicílico.
- Dosis inadecuada.
- Absorción reducida o incremento de su metabolismo.
Alteraciones en la unión a la COX1
- Ingestión conjunta con fármacos anti-inflamatorios no esteroides (por ejemplo ibuprofeno).
Otras fuentes de producción de tromboxano
- Biosíntesis de tromboxano por vías que no son bloqueadas por el ácido acetil salicílico (monocitos, macrófagos y células endoteliales).
Vías alternativas de activación plaquetaria
- Activación plaquetaria por vías que no son bloqueadas por el ácido acetil salicílico (por ejemplo, glóbulos rojos que inducen activación plaquetaria, estimulación del colágeno, ADP, epinefrina y receptores de trombina en las plaquetas).
- Incremento de la sensibilidad plaquetaria al colágeno y al ADP.
Incremento en el recambio de plaquetas
- Aumento en la producción de plaquetas por la médula ósea en respuesta al estrés (por ejemplo, después de cirugía de puentes aorto-coronarios).
Polimorfismo genético
- Polimorfismos de COX1, COX2 y del tromboxano A2.
- Polimorfismo de los receptores plaquetarios de glicoproteínas Ia/IIb/V/IX y IIb/IIIa, receptores del colágeno y factor de von Willebrand.
Pérdida del efecto antiplaquetario del ácido acetil salicílico por administración prolongada
- Taquifilaxia.
¿Cómo se realiza el diagnóstico de resistencia al ácido acetil salicílico?
Diagnóstico desde el punto de vista de laboratorio
La resistencia al ácido acetil salicílico puede determinarse por la medición de metabolitos estables como el TBXA2, el tromboxano B2 (plasma) y el 11 dihidrotromboxano B2 en la orina (Tabla 1).
Producción de tromboxano A2
La producción de TXBA2 puede estar determinada por sus metabolitos en el plasma o en la orina. Dado que la producción de tromboxano B2 (plasma), es altamente dependiente de la COX1 plaquetaria, éste se utiliza como medición de la inhibición plaquetaria a bajas dosis de ácido acetil salicílico (14).
Función plaquetaria dependiente de tromboxano
Los test de función plaquetaria que son dependientes de la producción de tromboxano plaquetario, incluyen agonistas que inducen la agregabilidad plaquetaria medidos por transmisión óptica o de luz. El tiempo de sangría es un test de función plaquetaria, dependiente, en parte, de la producción de tromboxano, y se usa muy poco por la alta dependencia del operador y porque que es pobremente reproducible. La agregometría óptica o por luz, mide la agregabilidad plaquetaria cuando las plaquetas en suspensión se someten a un agonista como por ejemplo el ADP, o el colágeno TBXA2, formando grupos de plaquetas (Figura 3).
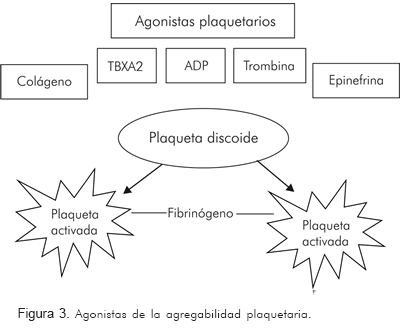
Este es el test que se ha mantenido a través del tiempo como «gold standard» para medir el efecto antiplaquetario del ácido acetil salicílico y aún es el más utilizado para determinar la función plaquetaria.
Sistema PFA-100
Se trata de un dispositivo perfeccionado del Thrombostat 400, originalmente desarrollado por Kratzer y Born. El analizador de función plaquetaria PFA-100 (Figura 4) simula, in vitro, la hemostasia primaria en sangre periférica mediante una lesión de membrana con colágeno/ADP o colágeno/epinefrina. Dicha membrana, cubierta por aproximadamente 50 mg de ADP y 10 mg de epinefrina, viene protegida en cartuchos perfectamente identificados con las iniciales COL/EPI o COL/ADP (Figura 4). El colágeno, la epinefrina y el ADP son, en condiciones fisiológicas in vivo, sustancias favorecedoras de la adhesión y agregación plaquetaria.

El sistema permite la colocación de los dos cartuchos o de uno sólo. Se requiere pipetear 8 mL de la muestra sanguínea no centrifugada en el orificio más pequeño del cartucho. EL total de la sangre debe procesarse en citrato sódico al 3,8%-3,2%, utilizado como anticoagulante (Figura 4).
El patrón típico en pacientes antiagregados con ácido acetil salicílico, del cual el sistema es muy específico, es: colágeno/epinefrina alargado y colágeno/ADP normal (27). La agregación plaquetaria medida por PFA-100 se ha correlaciona bien con los resultados obtenidos por agregometría óptica (28).
Diagnóstico clínico
La resistencia clínica al ácido acetil salicílico puede diagnosticarse clínicamente cuando ocurre un evento aterotrombótico isquémico en un paciente que tome una dosis terapéutica de ácido acetil salicílico (7-9, 29). El diagnóstico clínico de resistencia al ácido acetil salicílico es limitado debido a que es retrospectivo (hecho después del evento) y no es específico. Algunos autores piensan que es más apropiado clasificar a estos pacientes como falla en la respuesta terapéutica, que como una resistencia clínica a la terapia con ácido acetil salicílico(8).
Mediciones de agregación y activación plaquetaria, así como el tiempo de sangría, confirman la variabilidad de la respuesta antitrombótica a la terapia con ácido acetil salicílico.
Relevancia clínica
Estudios clínicos prospectivos demuestran que la disminución en la respuesta a la terapia con ácido acetil salicílico, incrementa el riesgo de enfermedad aterotrombótica (30-32). Varios estudios prospectivos validan la relación de resistencia al ácido acetil salicílico y el riesgo cardiovascular, y demuestan que la magnitud del riesgo puede ser importante (31, 32). Grundmann y colaboradores reportaron que en pacientes con eventos isquémicos previos, la incidencia de resistencia al ácido acetil salicílico era significativamente mayor (34%) en comparación con pacientes asintomáticos con enfermedad cerebrovascular conocida (33). Grotemeyer y colaboradores (34) reportaron 30% de incidencia de resistencia al ácido acetil salicílico entre pacientes que habían sufrido un evento isquémico cerebrovascular, después de la ingestión de 500 miligramos de ácido acetil salicílico. En el seguimiento a dos años los pacientes que no respondieron a este medicamento tuvieron un incremento diez veces mayor de recurrencia de eventos isquémicos, en comparación con los pacientes sensibles al mismo fármaco.
En pacientes con claudicación intermitente quienes se presentaron para angioplastia periférica, Mueller y colaboradores (35) reportaron 40% de resistencia al ácido acetil salicílico. En un seguimiento a 18 meses la resistencia al ácido acetil salicílico, se asoció con 87% de incremento del riesgo de reoclusión arterial.
En una cohorte del estudio Heart Outcome Prevention Evaluation (HOPE) Eikelboom y colaboradores (36) encontraron que el riesgo de infarto del miocardio, accidente isquémico cerebrovascular o muerte cardiovascular se incrementó con cada aumento de cuartil del 11 dihidro tromboxano B2 urinario. Aquellos en el cuartil superior tenían dos veces mayor riesgo para infarto del miocardio y tres veces mayor riesgo para muerte cardiovascular, cuando eran se compararon con aquellos en cuartiles más bajos. Chen y colaboradores (32) reportaron una asociación entre la resistencia al ácido acetil salicílico y la elevación de los niveles de la fracción MB de la creatinín kinasa, posterior a procedimientos de intervencionismo coronario. En este estudio de 151 pacientes con enfermedad coronaria estable, la incidencia de resistencia al ácido acetil salicílico medida por RPFA, fue de 19,2%.
Las observaciones de una gran cantidad de estudios clínicos aleatorizados, en pacientes con enfermedad coronaria quienes han experimentado algún proceso aterotrombótico y que se encontraban en terapia con ácido acetil salicílico, dan soporte a la validez de que el riesgo asociado es importante. Análisis posteriores del estudio ESSENCE (Efficacy and Safety of Subcutaneous Enoxaparin in Non-Q-wave Coronary Events) y el TIMI 11B (Thrombolysis In Myocardial Infarction 11B), reportaron que el uso previo de ácido acetil salicílico era un predictor independiente de incremento del riesgo cardiovascular en pacientes con síndrome coronario agudo (37).
Clopidogrel
Es una tienopiridina, química y farmacológicamente muy similar a la ticlopidina, que actúa modificando de manera selectiva e irreversible al receptor P2Y12 del ADP en la superficie de la plaqueta (Figura 5), e inhibe así la agregación plaquetaria (38). El ADP es un importante activador de la agregación plaquetaria y se encuentra en altas concentraciones en los gránulos de estos elementos formes. El ADP induce la agregación de las plaquetas al activar un receptor específico situado en la superficie externa de la membrana plaquetaria, lo que genera cambios en la concentración intracelular de calcio y la expresión y ensamblaje de receptores para el fibrinógeno en la superficie de la plaqueta (39).
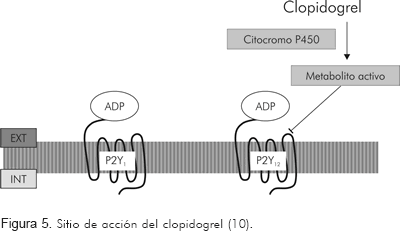
Farmacodinamia y farmacocinética
El clopidogrel es una prodroga que requiere oxidación por el sistema citocromo P450 hepático, para generar su metabolito activo (13). La unión del metabolito activo al receptor P2Y12 es irreversible y por tanto las plaquetas expuestas al clopidogrel se ven afectadas el resto de su vida. La máxima inhibición de agregación plaquetaria tras una dosis de carga del fármaco (375-400 mg) oscila entre 40% a 60% y se alcanza en unas dos a seis horas (40). En general, la agregación plaquetaria y el tiempo de sangría vuelven a los valores basales en los cinco días siguientes de la suspensión del tratamiento (40).
Cerca de 50% del fármaco se absorbe rápidamente en el tracto gastrointestinal y la cantidad que llega al plasma no se afecta por los alimentos ni por los antiácidos.
Resistencia al clopidogrel
El concepto de resistencia al clopidogrel emergió en la literatura médica con el fin de reflejar la falla de la inhibición plaquetaria in vitro, aunque su definición aún no se establece (13). Se propone que el término resistencia incluya a aquellos en quienes el clopidogrel no alcanza su efecto farmacológico, y la falla de la terapia se refleje en pacientes que cursan con eventos isquémicos recurrentes (41). La prevalencia de pacientes que no responden al clopidogrel se estima entre 4% a 30%, 24 horas después de su administración (4, 42-46).
Evaluación de la función plaquetaria
La función plaquetaria es medida in vitro, en la mayoría de los casos, mediante agregometría por transmisión de luz. Aunque este método es el más utilizado, posee algunas desventajas como reproducibilidad limitada y complejidad en la preparación de la muestra. Se dispone de otras técnicas pero son inadecuadas para monitorizar el efecto antiagregante del clopidogrel (47). El tiempo escogido para medir la agregación plaquetaria es de vital importancia, dado que 300 mg en bolo, pueden ejercer su efecto completo de antiagregación 24 horas después de su administración, en contraste con las cuatro horas en las que lo puede ejercer el bolo de 600 mg (48).
Mecanismos potenciales de resistencia al clopidogrel
Existen varios mecanismos de resistencia, los cuales se clasifican en intrínsecos y extrínsecos (13) (Tabla 2). Además, puede existir variabilidad en la absorción asociada con dosis bajas del medicamento (49).
Los mecanismos extrínsecos que reflejan una biodisponibilidad reducida del clopidogrel, pueden incluir incumplimiento de la terapia, baja dosis e interacción con otros medicamentos que afecten la biotransformación del clopidogrel en su metabolito activo (41). Los posibles mecanismos intrínsecos pueden incluir polimorfismo del receptor P2Y12 y del sistema citocromo P450.
Interacción con otros medicamentos
Cualquier medicamento que inhiba o sea sustrato del sistema citocromo P450 puede bloquear potencialmente la conversión del clopidogrel en su metabolito activo. Entre estos medicamentos figuran los inhibidores de la hidroximetil glutaril CoA reductasa, comúnmente denominados estatinas (50). Existen análisis que sugieren la posibilidad de interacción entre atorvastatina y clopidogrel (51). El análisis del estudio CREDO encontró que esta interacción no fue estadísticamente significativa y el beneficio de clopidogrel fue similar con todas las estatinas a pesar de su interacción metabólica (52). Otros datos provenientes del estudio INTERACTION (Interaction of Atorvastatin and Clopidogrel) (53), demuestran que las estatinas, incluyendo la atorvastatina, no interfieren con la agregación plaquetaria mediada por clopidogrel. Adicionalmente, estudios realizados con mayores dosis de clopidogrel y estatinas, demuestran carencia de interacción entre estos agentes (54).
Entidades clínicas
Un análisis reciente correlacionó el nivel de angina según la clasificación de Braunwald, con la inhibición plaquetaria con clopidogrel. Se encontró que los pacientes con una clasificación más severa de angina, tenían una inhibición plaquetaria más baja (55). Matetzki y colaboradores (56) analizaron 60 pacientes con infarto agudo del miocardio con elevación del segmento ST quienes fueron llevados a angioplastia primaria y recibieron clopidogrel en bolo 300 mg y luego 75 mg por tres meses. Los pacientes se dividieron en cuartiles de acuerdo con la respuesta al clopidogrel medida por agregación plaquetaria inducida por ADP. Aquellos que mostraron menor respuesta al clopidogrel presentaron 25% más probabilidad de evento recurrente cardiovascular durante los seis meses de seguimiento. Además, se ha encontrado mayor probabilidad de trombosis subaguda del stent, en pacientes con angina estable sometidos a angioplastia coronaria que no responden a terapias con clopidogrel (43). Más grave aún, Lev y colaboradores (57) realizaron un estudio, recientemente publicado, con 150 pacientes sometidos a intervención coronaria percutánea y en quienes midieron la agregabilidad plaquetaria tanto para ácido acetil salicílico como para clopidogrel. Encontraron que 12,7% de los pacientes eran resistentes al ácido acetil salicílico y 24% al clopidogrel; además, 6% de los pacientes fueron resistentes a ambos medicamentos y presentaron niveles elevados de CKMB después de la intervención percutánea, lo cual representa alto riesgo para eventos trombóticos posteriores al procedimiento.
Variabilidad en la respuesta al clopidogrel
Los mecanismos que conllevan a variabilidad en la respuesta al tratamiento con clopidogrel, no están dilucidados y al parecer, como ocurre con el ácido acetil salicílico, las cusas son multifactoriales (58). La alta reactividad plaquetaria diagnosticada antes del tratamiento, puede contribuir a una reducción del efecto antiplaquetario del clopidogrel. Este incremento en la reactividad plaquetaria basal puede observarse en situaciones clínicas específicas como síndromes coronarios agudos, incremento en el índice de masa corporal y diabetes mellitus, en particular los insulino-dependientes (41-58). Otros factores clínicos que pueden conducir a una reducción en el efecto del clopidogrel son el incumplimiento terapéutico completo por parte del paciente, la dosis inadecuada, las diferencias en el grado de absorción y los niveles del metabolito activo.
Medidas terapéuticas para la resistencia al ácido acetil salicílico y al clopidogrel
En la actualidad no existe evidencia que indique que aumentar la dosis de ácido acetil salicílico pueda ser útil (6, 59); además, es preciso recordar que el aumento de dosis puede producir mayores efectos secundarios no deseados sobre todo en el tracto gastrointestinal (60) y dicho aumento cambia al efecto analgésico y antipirético y no antiagregante como se explicó anteriormente. Los médicos deben cerciorarse de que el paciente tome la dosis apropiada de ambos medicamentos y que presente niveles adecuados de colesterol y glucosa para reducir la reactividad plaquetaria. La adición de clopidogrel al ácido acetil salicílico es lógica debido a sus diferentes mecanismos de acción. El estudio CAPRIE (The clopidogrel vs. aspirin in Patients at Risk for Ischaemic Events) (61) reveló una modesta superioridad del clopidogrel sobre el ácido acetil salicílico como monoterapia, y también mostró un aumento del beneficio en pacientes de alto riesgo.
En cuanto al clopidogrel, el aumento de dosis en bolo demuestra una inhibición plaquetaria más rápida con mejoría en eventos clínicos, a diferencia de resultados obtenidos con el aumento de dosis de ácido acetil salicílico. El estudio ARMIDA 2 (The Antiplatelet Therapy for Reduction of Myocardial Damage during Angioplasty) mostró el beneficio de 600 mg de clopidogrel comparado con 300 mg (dosis bolo) en reducir el infarto periprocedimiento en pacientes sometidos a intervención coronaria percutánea (62). La utilidad en el incremento de la dosis de clopidogrel, se evaluó en el estudio ALBION (Loading Dose of Clopidogrel to Blunt Platelet Activation, Inflammation and Ongoing Necrosis) donde se compararon dosis de 300, 600 y 900 mg de clopidogrel en bolo, en pacientes con síndrome coronario agudo sin elevación del segmento ST. La dosis de 900 mg estuvo asociada con una inhibición plaquetaria más rápida, con tendencia a una disminución en los niveles de marcadores de necrosis miocárdica y efectos cardiacos mayores adversos, aunque estos resultados no tuvieron significancia estadística (63).
Basándose en la evidencia actual, las guías del Colegio Americano de Cardiología, sugieren (indicación clase IIb y nivel de evidencia C), que sólo los pacientes en quienes la trombosis del stent pueda ser un evento catastrófico o letal (tronco izquierdo no protegido, bifurcación de tronco izquierdo y único vaso permeable) la dosis de clopidogrel puede aumentarse a 150 mg/día si se demuestra una inhibición de la agregación plaquetaria menor de 50% (64).
Perspectivas futuras y nuevos fármacos
Está en curso el estudio RESISTOR (Research Evaluation to Study Individuals who Show Thromboxane or P2Y12 Receptor Resistance), el cual evaluará a los pacientes sensibles al ácido acetil salicílico y al clopidogrel que serán llevados a angioplastia coronaria y posteriormente recibirán heparina no fraccionada o un inhibidor de la glicoproteína IIb/IIIa (eftifibatide), con el objetivo de estudiar a fondo la resistencia al inhibidor P2Y12. Han surgido nuevos inhibidores del receptor P2Y12 como el prasugrel, el cual, en estudios preclínicos, parece ser más potente y tener un inicio de acción más rápido que el clopidogrel (65). Estudios fase 1 en humanos sanos sin previa ingestión de ácido acetil salicílico, mostraron una mejor inhibición plaquetaria con 60 mg de prasugrel que con 300 mg de clopidogrel (66); así mismo, una dosis de mantenimiento de 10 mg de prasugrel mostró mayor inhibición de la agregación plaquetaria que 75 mg de clopidogrel (67). Se publicaron los resultados del JUMBO TIMI–26 (Join Utilization of Medication to Block Platelets Optimally), un estudio fase II de seguridad, con 904 pacientes en el que se compara al clopidogrel con tres regímenes de dosis de prasugrel. Ambos medicamentos mostraron tasas muy bajas de sangrado y hubo menor incidencia de efectos cardiacos adversos con prasugrel, aunque este hallazgo no mostró significancia estadística (68). Está en curso el TRITON TIMI 38 (Trial to Assess Improvements in Therapeutics Outcomes by Optimizing Platelet Inhibition with Prasugrel Thombolysis in Myocardial Infaction 38), que es un estudio fase III, que compara clopidogrel vs. prasugrel, y tiene la meta de incluir 13.000 pacientes con síndrome coronario agudo. Es el primer estudio a gran escala para determinar si los niveles altos de inhibición plaquetaria conllevan mejores resultados clínicos (69).
Existen otros fármacos que se encuentran en investigación como el cangrelor, un antagonista directo no tienopiridínico del receptor P2Y12, de uso intravenoso, el cual tiene un potente efecto antiagregante plaquetario (mayor que el clopidogrel), una vida plasmática corta y no necesita activación hepática (70, 71) y el ADZ6140, un antagonista del receptor P2Y12 de administración oral, que tampoco requiere metabolismo hepático y produce un bloqueo de la agregación plaquetaria más completo que el clopidogrel (72).
Conclusión
Pese a que el término resistencia al ácido acetil salicílico y al clopidogrel es muy usado, su definición es vaga. Se emplea para denotar la falla de ambos medicamentos en ejercer su efecto de inhibición plaquetaria. Existe evidencia substancial que sugiere que un porcentaje importante (5% a 40%) de individuos que toman ácido acetil salicílico y (4% a 30%) de los que ingieren clopidogrel, han demostrado resistencia a sus efectos antitrombóticos, lo que conlleva un incremento en el riesgo de enfermedad aterotrombótica incluyendo muerte, infarto del miocardio y enfermedad cerebrovascular.
Existen diversos factores tanto intrínsecos como extrínsecos, así como una variabilidad individual y multifactorial que puede contribuir a que se desarrolle resistencia tanto al clopidogrel como al ácido acetil salicílico; más aun, se identificaron grupos de pacientes resistentes a ambos medicamentos, los cuales están en muy alto riesgo, sobre todo sin son llevados a intervencionismo coronario. Los nuevos fármacos inhibidores del receptor P2Y12 se vislumbran como una posible arma terapéutica para pacientes resistentes y de alto riesgo. Existen entidades clínicas que pueden predisponer a cierto grado de resistencia al ácido acetil salicílico y al clopidogrel; estos casos deben tener un estricto seguimiento para su identificación y toma de medidas terapéuticas adecuadas.
Los estudios clínicos corroboran que el aumento de la dosis de clopidogrel se relaciona con una mayor y más rápida inhibición de la agregabilidad plaquetaria con mejoría en eventos clínicos, resultados que no han sido similares con el ácido acetil salicílico. En un futuro cercano, medir la función plaquetaria en pacientes con enfermedad cardiovascular, llegará a ser un objetivo de primera línea y de fácil realización para identificar pacientes con riesgo elevado. Se necesitan estudios adicionales a gran escala, como los que se llevan a cabo en la actualidad, para determinar las consecuencias clínicas de los hallazgos de laboratorio en relación con los pacientes que desarrollen resistencia al ácido acetil salicílico, al clopidogrel o a ambos.
Bibliografía
1. Fuster V, Badimon L, Badimon JJ, Chesebro JH. The pathogenesis of coronary artery disease and the acute coronary syndromes. N Engl J Med 1992; 326: 310-318. [ Links ]
2. Libby P. Molecular bases of the acute coronary syndromes. Circulation 1995; 91: 2844-2850. [ Links ]
3. Gum PA, Kottke-Marchant K, Poggio ED, et al. Profile and prevalence of aspirin resistance in patients with cardiovascular disease. Am J Cardiol 2001; 88: 230-35. [ Links ]
4. Gurbel PA, Bliden KP, Hiatt BL, O‘Connor CM. Clopidogrel for coronary stenting: response variability, drug resistance and the effect of pre-treatment platelet reactivity. Circulation 2003; 107: 2908-13. [ Links ]
5. Gurbel PA, Bliden KP. Durability of platelet inhibition by clopidogrel. Am J Cardiol 2003; 91: 1123-5. [ Links ]
6. Wang TH, Bhatt DL, Topol EJ. Aspirin resistance: an emerging clinical entity. Eur Heart J 2005; 93: 02-08. [ Links ]
7. Bhat DL. Aspirin resistance: more than just a laboratory curiosity. J Am Coll Cardiol 2004; 43: 1127-29. [ Links ]
8. Hankey GJ, Eikelboom JW. Aspirin resistance. Lancet 2006; 367: 606-617. [ Links ]
9. Mason P J, Jacobs AK, Freedman JE. Aspirin resistance and atherothrombotic disease. J Am Coll Cardiol 2005; 46: 986-93. [ Links ]
10. Cattaneo M. Aspirin and clopidogrel: efficacy, and the issue of drug resistance. Arterioscler Thromb Vasc Biol 2004; 24: 1980-87. [ Links ]
11. Buchanan MR, Brister SJ. Individual variation in the effect of ASA on platelet function: implications for the use of ASA clinically. Can J Cardiol 1995; 11: 221-7. [ Links ]
12. Roth GL, Carverley DC. Aspirin, platetlets and thrombosis theory and practice. Blood 1994; 83: 885-98. [ Links ]
13. Nguyen TA, Diodaty JG, Pharand C. Resistance to clopidogrel: A review of the evidence. J Am Coll Cardiol 2005; 45: 1157-64. [ Links ]
14. Patrono C. Aspirin resistance: definition, mechanism and clinical read-outs. J Thromb Haemost 2003; 1: 1710-3. [ Links ]
15. Patrono C, Garcia L, Landolfi R, Baigent C. Low–dose aspirin for the prevention of atherothrombosis. N Engl J Med 2005; 353: 2373-83. [ Links ]
16. Pedersen AK, FitzGerald GA. Dose related kinetics of aspirin: presystemic acetylation of platetlet cyclooxygenase. N Engl J Med 1984; 311: 1206-11. [ Links ]
17. Patrono C, Coller B, FitzGerald GA, Hirsh J, Roth G. Platelet-active drugs: the relationships among dose, effectiveness, and side effects: The Seventh ACCP Conference on Antithrombotic and Thrombolitic Therapy. Chest 2004; 126: (Suppl): 234S-246S. [ Links ]
18. Belton O, Byrne D, Kearney D, Leahy A, Fitzgerald DJ. Cyclooxigenasa-1 and 2- dependent prostacyclin formation in patients with atherosclerosis. Circulation 2000; 102: 840-5. [ Links ]
19. Catella-Lawson F, Reilly MP, Kapoor SC, et al. Cyclooxigenasa inhibitors and the antiplatelet effects of aspirin. N Engl J Med 2001; 345: 1809-17. [ Links ]
20. Capone MI, Sciulli MG, Tacconelli S, et al. Pharmacodynamic interaction of naproxen with low-dose aspirin in healthy subjets. J Am Coll Cardiol 2005; 45: 1295-301. [ Links ]
21. Patrono C, Coller B, Dallen JE, et al. Platelet active drugs: the relationship among dose, effectiveness, and side effects. Chest 2001; 119: 39S-63S. [ Links ]
22. Califf RM, DeLong ER, Ostbye T, Muhlbaier LH, Chen A, LaPointe NA, et al. Underuse of aspirin in a referral population with documented coronary artery disease. Am J Cardiol 2002; 89: 653-61. [ Links ]
23. Serabruany VL, Malinin AL, Jerome SD, Lowry DR, Morgan AW, Sane DC, et al. Effects of clopidogrel and aspirin combination versus aspirin alone on platelet aggregation and major receptor expression in patients with heart failure (PLUTO-CHF) trial. Am Heart J 2003; 146: 713-20. [ Links ]
24. Undas A, Brummel K, Musial J, Mann KG, Szczeklik A. Pl (A2) polimorfism of beta 3 integrins is associated with enhance thrombin generation and impaired antithrombotic action of aspirin at the site of microvascular injury. Circulation 2001; 104: 2666-72. [ Links ]
25. Pontiggia L, Lassila R, Pederiva S, Schmid HR, Burger M, Beer JH. Increased platelet-colagen interaction associated with double homozygosity for receptor polimorfism of platelet GPIa and GP IIIa. Arterioscler Thromb Vasc Biol 2002; 22: 2093-98. [ Links ]
26. Harrison P, Robinson MSC, et al. Performance of the Platelet Function Analyser (PFA- 100) in testing abnormalities of primary haemostasis. Blood Coagulation and Fibrinolisis 1999; 10: 25-31. [ Links ]
27. Kottke-Marchant K, Power JB et al. The effect of antiplatelet drugs, heparin and preanalytcal variables on platelet function detected by the platelet function analyser (PFA-100). Clin Appl thrombosis/Hemostasis 1999; 5: 122-30. [ Links ]
28. Mammen EF, Comp PC, Gosselin R et al. PFA -100 system: a new method for assessment of platelet disfunction. Semin Thromb Haemost 1998; 24: 195-202. [ Links ]
29. Bhatt D, Topol EJ. Scientific and therapeutic advance in antiplatelet therapy. Nature Rev 2003; 2: 15-28. [ Links ]
30. Grotemeyer KH, Scharafinski HW, Husstedt IW. Two years follow-up of aspirin responder and aspirin non responder. A pilot study including 180 post-stroke patients. Thromb Res 1993; 71: 397-403. [ Links ]
31. Gum PA, Kottke-Marchant K, Welsh PA et al. A prospective blinded determination of the natural history of aspirin resistance among stable patients with cardiovascular disease. J Am Coll Cardiol 2003; 41: 961-5. [ Links ]
32. Chen W-H, Lee P-Y, Ng W et al. Aspirin resistance is associated with a high incidence of myonecrosis after non-urgent percutaneous coronary intervention despite clopidogrel pre-treatment. J Am Coll Cardiol 2004; 43: 1122-6. [ Links ]
33. Grundmann KJK, Kleine B, Dichgans J, et al. Aspirin non responder status in patients with recurrent cerebral ischaemic attacks. J Neurol 2003; 250: 63-6. [ Links ]
34. Grotemeyer KH. Effects of acetylsalicylic acid in stroke patients. Evidence of nonresponders in a subpopulation of treated patients. Thromb Res 1991; 63: 587-93. [ Links ]
35. Mueller MR, Salat A, Stangl P, et al. Variable platelet response to low-dose ASA and the risk of limb deterioration in patients submitted to peripheral arterial angioplasty. Thromb Haemost 1997; 78: 1003-7. [ Links ]
36. Eikelboom JW, Hirsh J, Weitz JI, Johnston M, Yi Q, Yusuf S. Aspirin resistance thromboxane biosyntesis and the risk of myocardial infarction, stoke or cardiovascular death in patients at high risk for cardiovascular events. Circulation 2002; 105: 1650-55. [ Links ]
37. Antman EM, Cohen M, Bernink PJ, et al. The TIMI risk score for unstable angina / non-ST elevation MI: a method for prognostication and therapeutics decision making. JAMA 2000; 284: 835-42. [ Links ]
38. Savi P, Heilmann E, Nurden P. Clopidogrel: an antithrombotic drug acting on ADP dependent activation pathway of human platelet. Thromb Haemost 1996; 2: 35-42. [ Links ]
39. Savi P, Pereillo JM, Uzabiaga MF et al. Identification and biological activity of the active metabolite of clopidogrel. Thromb Haemost 2000; 84: 891-6. [ Links ]
40. Quinn MJ, Fitzgerard DJ. Ticlopidine and clopidogrel. Circulation 1999; 100: 1667-72. [ Links ]
41. Wiviott SD, Antmann EM. Clopidogrel resistance: a new chapter in a fast-moving story. Circulation 2004; 109: 3064-7. [ Links ]
42. Jeremo P, Lindahl TL, Fransson SG, Richter A. Individual variations of platelet inhibition after loading dose of clopidogrel. J Intern Med 2002; 252: 233-8. [ Links ]
43. Muller I, Besta F, Schulz C, Massberg S, Schoring A, Gawaz M. Prevalence of clopidogrel non-responders among patients with stable angina pectoris scheduled for elective coronary stent placement. Thromb Haemost 2003; 91: 1123-25. [ Links ]
44. Mobley JE, Bresee SJ, Wortham DC, Craft RM, Snider CC, Carrol RC. Frequency of non-responder antiplatelet activity of clopidogrel during pre-treatment for cardiac catheterization. Am J Cardiol 2004; 93: 456-8. [ Links ]
45. Angiolillo DJ, Fernandez-Ortiz A, Bernardo E, et al. Platelet aggregation according to body mass index in patients undergoing coronary stenting should clopidogrel loading dose be adjusted? J Invasive Cardiol 2004; 16: 169-74. [ Links ]
46. Serebruany VL, Steinhubl SR, Berger PB, Malinin AI, Bhatt DL, Topol EJ. Variability in platelet responsiveness to clopidogrel among 544 individuals. J Am Coll Cardiol 2005; 45: 246-51. [ Links ]
47. Michelson AD. Platelet function testing in cardiovascular diseases. Circulation 2004; 110: 489-93. [ Links ]
48. Muller I, Seyfarth M, Rudiger S, et al. Effect of a high loading dose of clopidogrel on platelet function in patients undergoing coronary stent placement. Heart 2001; 85: 92-3. [ Links ]
49. Taubert D, Kastrati A, Harlfingers S, Gorchakova O,Lazar A, von Beckerat N, et al. Pharmacokinetics of clopidogrel after administration in high loading dose. Thromb Haemost 2004; 92: 311-316. [ Links ]
50. Lau WC, Gurbel PA, Watkins PB, Neer CJ, Hoops AS, Carville DG, et al. Contribution of hepatic citocrome P450 3A4 metabolic activity to the phenomenon of clopidogrel resistance. Circulation 2004; 109: 166-71. [ Links ]
51. Lau WC, Waskell LA, Watkins PB, Neer CJ, Horowitz K, Hoop AS, et al. Atorvastatin reduces the ability of clopidogrel to inhibit platelet aggregation: a new drug-drug interaction trial. Circulation 2003; 108: 921-24. [ Links ]
52. Saw J, Steinhubl SR, Berger PB, Kereakis DJ, Serebruany VL, Brennan D, Topol EJ. Lack of adverse clopidogrel-atorvastatin clinical interaction from secondary analysis of a randomised, placebo-controlled clopidogrel trial. Circulation 2003; 107: 332-37. [ Links ]
53. Serebruany VL, Midei MG, Malinin AL, Oshrine BR, Lowry DR, Sane DC, et al. Absence of interaction between atorvastatin or others statins and clopidogrel: results of interaction study. Arch Intern Med 2004; 164: 2051-57. [ Links ]
54. Gorchakova O, von Beckerat N, Gawaz M, Mocz A, Joost A, Schomig A, et al. Antiplatelet effect of a 600 mg loading dose of clopidogrel are not attenuated in patients receiving atorvastatin or sinvastatin for at least 4 weeks prior to coronary artery stenting. Eur Heart J 2004; 25: 1898-1902. [ Links ]
55. Soffer D, Moussa I, Harjai K, Boura JA, Dixon SR, Grines CLet al. Impact of angina class on inhibition of platelet aggregation following clopidogrel loading in patients undergoing coronary interventions: do we need more aggressive dosing regimens in unstable angina? Catheter Cardiovasc Interv 2003; 59: 21-25. [ Links ]
56. Matetzky S, Shenkman B, Guetta V, Shechter M, Biernart R, Goldenberg I, et al Clopidogrel resistance is associated with increased risk of recurrent atherothrombotic events in patients with acute myocardial infarction. Circulation 2004; 109: 3171-3175. [ Links ]
57. Lev EI, Patel TR, Maresh KJ, Guthikonda S, Granada J, De Lao T, et al. Aspirin and clopidogrel drug response in patients undergoing percutaneous coronary intervention. J Am Coll cardiol 2006; 47: 27-33. [ Links ]
58. Angiolillo D, Ortiz A, Bernardo E, Macaya C, Bass T, Costa M. Variability in individual responsiveness to clopidogrel. J Am Coll Cardiol 2007; 49: 1505-16. [ Links ]
59. Antithrombotic trialist collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324: 71-86. [ Links ]
60. Peters RJG, Metha SR, Fox KAA, Zhao F, Lewis BS, Kopecky SL, et al, for the Clopidogrel in Unstable Angina to Prevent Recurrent Events (CURE) trial investigators. Effects of aspirin dose when used alone or in combination with clopidogrel in patients with acute coronary syndromes: observations from the clopidogrel in Unstable Angina to Prevent Recurrent Events (CURE) study. Circulation 2003; 108: 1682-1687. [ Links ]
61. CAPRIE Steering committee. A randomised, blinded trial of clopidogrel versus aspirin in patients at risk for ischaemic events. Lancet 1996; 348: 1329-1339. [ Links ]
62. Patti G, Colonna G, Pasceri V, Pepe L, Montinaro A, Di Sciascio G. Randomised trial of high loading dose of clopidogrel for reduction of periprocedural myocardial infarction in patients undergoing coronary interventions. Results from the ARMIDA-2 (Antiplatelet Therapy for Reduction of Myocardial Damage during Angioplasty) Study. Circulation 2005; 111: 2099-2106. [ Links ]
63. Montalescot G, Sideris G, Meuleman C, et al. A randomised comparison of high clopidogrel loading doses in patients with non-ST-segment elevation acute coronary syndrome. The ALBION (Assessment of the Best Loading Dose of Clopidogrel to Blunt Platelet Activation, Inflammation and Ongoing Necrosis trial. J Am Coll Cardiol 2006; 48: 931-8. [ Links ]
64. Smith SC, Jr., Feldman TE, Hirshfeld JW Jr., et al. ACC /AHA/ SCAI 2005 guidelines update for percutaneous coronary intervention: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (ACC/AHA/SCAI Writing Committee to Update the 2001 Guidelines for percutaneous Coronary Intervention). J Am Coll Cardiol 2006; 47: e1-121. [ Links ]
65. Sugidashi A, Asai F, Ogawa T, Inoue T, Koike H. The in vivo pharmacological profile of CS-747 a novel antiplatelet agent with platelet ADP receptor antagonist properties. Br J Pharmacol. 2000; 129: 1439-46. [ Links ]
66. Brandt JT, Payne CD, Weerakkody G, Behoudek BD, Naganuma H, Wiviott SD, Winters KJ. Superior responder rate for inhibition of platelet aggregation with 60 mg loading dose of prasugrel (CS-747, LY640315) compared with 300 mg loading dose of clopidogrel. J Am Coll Cardiol. 2005; 45 (suppl A): 87A. Abstract. [ Links ]
67. Asay F, Jakubowsky JA, Hirota T, Matsushima N, Naganuma H, Freestne S. A comparison of prasugrel (CS-747, LY640315) with clopidogrel on platelet function in healthy male volunteers. J Am Coll Cardiol. 2005; 45 (suppl A): 87A. Abstract. [ Links ]
68. Wiviott SD, Antman EM, Winters KJ, Weerakkody G, Mutphy SA, Behounek BD, et al. Randomized comparison of prasugrel (CS-747, LY640315), a novel thienopiridine P2Y12 antagonist, with clopidogrel in percutaneous coronary intervention: results of the Join Utilization of Medication to Block Platelets Optimally (JUMBO)-TIMI 26 trial. Circulation 2005; 111: 3366-73. [ Links ]
69. Wiviott SD, Antman EM, Gibson CM, Montalescot G, Reisemeyer J, Weerakkody G, et al. Evaluation of prasugrel compared with clopidogrel in patients with acute coronary syndromes: design and rationale for the Trial to Assess Improvements in Therapeutics Outcomes by Optimizing Platelet Inhibition with Prasugrel Thombolysis in Myocardial Infarction 38 (TRITON TIMI 38). Am Heart J 2006; 152: 625-35. [ Links ]
70. Storey F. The P2Y12 receptor as a therapeutic target in cardiovascular disease. Platelets 2001; 12: 197-209. [ Links ]
71. Bhatt DL, Topol EJ. Scientific and therapeutic advances in antiplatelet therapy. Nat Rev Drug Discov 2003; 2: 15-28. [ Links ]
72. Husted S, Emanuelsson H, Heptinstall S, Sandset PM, Wickens M, Peters G. Pharmacodynamic, pharmacokinetics and safety of the oral reversible P2Y12 antagonist AZD6140 with aspirin in patients with atherosclerosis: a double-blind comparison to clopidogrel with aspirin. Eur Heart J 2006; 27: 1038-47. [ Links ]

