










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Cardiología
Print version ISSN 0120-5633
Rev. Colom. Cardiol. vol.16 no.3 Bogota May/June 2009
Universidad CES, Medellín, Colombia.
Correspondencia: Mauricio Duque, MD. Universidad CES, Calle 10a No. 22-04, Medellín, Colombia. Teléfono: 3121986. Correo electrónico: mauricioduque@une.net.co
Recibido: 30/13/2009. Aceptado: 16/06/2009.
La amiloidosis cardiaca es una manifestación de un grupo de enfermedades sistémicas que en conjunto se conocen como amiloidosis. Se considera una causa importante de las enfermedades infiltrativas que pueden ser responsables tanto del compromiso cardiaco como de otros órganos. Dado el avance en el entendimiento de la fisiopatología de la enfermedad, el reconocimiento de sus causas primarias y secundarias, y de las nuevas opciones terapéuticas, se describe un caso típico del compromiso cardiaco y posteriormente se hace una revisión del tema que abarca la mayoría de los tópicos que guardan relación con la amiloidosis primaria con compromiso cardiaco.
Palabras clave: cardiomiopatía restrictiva, amiloidosis cardiaca, falla cardiaca.
Cardiac amyloidosis is a manifestation of a group of systemic diseases, known as amyloidosis. It is considered an important cause of infiltrative diseases that may be responsible for both the heart and other organs' involvement. Given the progress in the understanding of the pathophysiology of the disease, the recognition of its primary and secondary causes and the new therapeutic options, a typical case of cardiac involvement is described and a review of the subject that covers the great majority of topics related to primary amyloidosis with cardiac involvement is made.
Key words: restrictive cardiomyopathy, cardiac amyloidosis, heart failure.
Caso
Hombre de 56 años de edad, con antecedente de hipertensión, quien a mediados de 2006 tuvo tos no productiva. Recibía inhibidores de la enzima convertidora de angiotensina y a éstos se les atribuyó el síntoma, por lo cual se suspendieron aunque no se observó mejoría. La radiografía de tórax fue normal. Persistió con la tos y aparecieron otros síntomas como disnea de moderados esfuerzos, fatiga y pérdida objetiva de peso de 20 kg, por lo cual recibió valoración por neumología. Se hizo ecocardiograma, gammagrafía de ventilación/perfusión y Doppler venoso de miembros inferiores los cuales fueron normales. Al siguiente mes se le realizó una nueva radiografía de tórax que mostró derrame pleural bilateral y una ecografía que confirmó el hallazgo asociado a atelectasias pasivas de ambos segmentos basales y posteriores de ambos lóbulos inferiores.
Se remitió al servicio de gastroenterología para estudio de posible reflujo como causa de la tos y la disnea, y allí se le realizó una pH-metría esofágica de dos sensores que reportó reflujo gastroesofágico grado I y aclaración esofágica levemente disminuida de predominio nocturno. Índice de síntomas negativo para tos, probabilidad de asociación al síntoma negativo para la tos y nulo compromiso del esófago proximal. Posteriormente se realizó endoscopia digestiva superior e inferior en la que se confirmó el diagnóstico de reflujo gastroesofágico y consecuente esofagitis leve y gastritis antral eritematosa. Se informó en las biopsias tomadas como gastritis crónica activa severa con presencia de Helicobacter pylori; no se observaron cambios compatibles con metaplasia ni malignidad. En el estudio del tracto digestivo inferior se documentaron lesiones tipo pólipo que fueron resecadas e informadas como adenomas tubulovellosos con displasia de bajo grado y escasos focos de displasia de alto grado, uno de ellos hiperplásico y otro adenoma tubular con displasia de bajo grado. No se realizaron tinciones especiales para estudio de enfermedad de depósito. La ecografía abdominal total fue normal.
Para septiembre de 2007 el paciente consultó nuevamente por progresión de la disnea y la fatiga sin evidencia clara de la causa de las mismas y debido al deterioro de su clase funcional, se le realizó un nuevo ecocardiograma que reportó:
- Ventrículo izquierdo de tamaño normal con hipertrofia concéntrica severa, disminución de la cavidad ventricular, sin trastornos segmentarios en la contractilidad.
- Fracción de eyección mayor de 60%.
- Disfunción diastólica tipo III (patrón restrictivo).
- Ventrículo derecho de tamaño normal con hipertrofia de la pared libre y función normal. Presión sistólica de la arteria pulmonar de 55 a 60 mm Hg.
Se llevó a coronariografía, donde se documentó lesión crítica en la unión del tercio medio y el distal de la coronaria derecha. Se implantó un stent convencional con resultado angiográfico satisfactorio. Posteriormente se dio de alta con manejo médico.
Consultó una vez más por persistencia de sus síntomas y presencia de edema en miembros inferiores. Se retomó el caso y para el estudio de la hipertensión pulmonar se realizó angio-TAC de tórax con sospecha de trombo-embolismo recurrente, el cual fue negativo para tal diagnóstico y para lesiones en el parénquima pulmonar. Se evidenció nuevamente la presencia de derrame pleural bilateral que fue sometido a estudio, reportándose negativo para malignidad. Se dio de alta con manejo médico.
Reingresó dos semanas después por empeoramiento de sus síntomas y progresión del edema en miembros inferiores. Fue evaluado por cirugía por hallazgo de ascitis y síndrome edematoso. Se programó para laparoscopia y biopsia, y por presentar prolongación de los tiempos de coagulación secundaria a una hepatomegalia congestiva y recibir aspirina se esperó hasta compensarse de la misma. Una semana más tarde se realizó dicho procedimiento sin complicaciones. El informe reportó líquido ascítico negativo para malignidad, cultivos microbiológicos negativos, y los distintos análisis lo hacían incompatible con hipertensión portal. Se discutió en staff de cardiología y dado lo reciente de la coronariografía y por recibir ASA/clopidogrel se consideró prudente no realizar biopsia endomiocárdica. Además se decidió no iniciar antagonistas del calcio sin realizar un test de vaso-reactividad pulmonar y adicionar un beta-bloqueador cardio-selectivo tipo nevibolol sumado al manejo de su enfermedad coronaria y falla cardiaca secundaria a la disfunción diastólica. Adicionalmente, se inició sildenafil y de acuerdo con la respuesta clínica se pensaría en agregar antagonistas de la endotelina. Durante este periodo se evaluaron: perfil lipídico, metabólico y hematológico, y función tiroidea y renal, los cuales fueron normales.
Fue valorado en nuestro servicio, y allí relató, luego de 18 meses de evolución, que a pesar de los múltiples estudios e intervenciones su condición clínica había empeorado en forma dramática. Se realizó electrocardiograma y se documentó bradicardia sinusal y bajo voltaje generalizado (Figura 1).
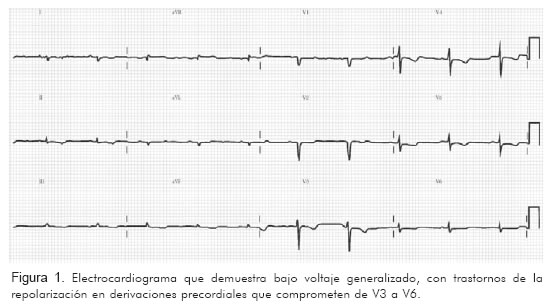
Con la historia clínica descrita y el electrocardiograma se pensó en enfermedad infiltrativa y se tomó biopsia endomiocárdica, la cual se realizó sin complicaciones. El informe de patología (Figura 2) indicó:
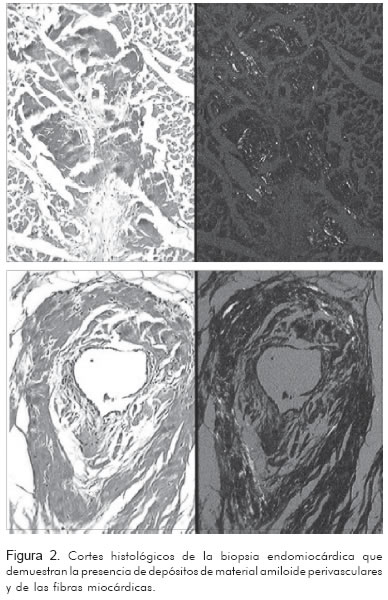
- Coloraciones de cristal violeta y rojo Congo que muestran positividad en la forma de depósitos delicados de material amiloide alrededor de los vasos sanguíneos y fibras miocárdicas individuales.
- Imagen histológica muy sugestiva de amiloidosis.
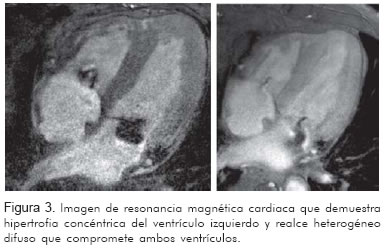
Se realizó resonancia nuclear magnética cardiaca que mostró hipertrofia concéntrica del ventrículo izquierdo con alteración en la función diastólica. La pared septal midió en sístole 22 mm y en diástole 17 mm y una fracción de expulsión de 56%. En las imágenes de realce tardío se evidenció un realce heterogéneo difuso que comprometía tanto el ventrículo izquierdo como el derecho e incluso las aurículas, compatible con patrón infiltrativo tipo amiloidosis (Figura 3).
Fue valorado por hematología para estudio y manejo. Se realizó mielograma, biopsia de médula ósea y electroforesis de proteínas en suero y orina.
Los resultados de estos estudios fueron los siguientes:
· Biopsia de médula ósea:
- Adecuada diferenciación y población de las tres líneas celulares.
- Negativa para depósito amiloide.
· Revisión de bloques de parafina de epiplón, peritoneo e hígado:
- Negativos para depósito amiloide.
Se sospechó la presencia de amiloidosis primaria con compromiso cardiaco y se inició manejo con ciclofosfamida, talidomida y prednisolona.
Dada la poca experiencia que se tiene en el país para el estudio y manejo de esta condición, los estudios se remitieron a la clínica Mayo en Rochester conocida por ser centro de remisión en los Estados Unidos para el estudio y manejo de esta patología.
En comunicación directa, el Doctor Morie A. Gerts, hematólogo especialista en amiloidosis y mieloma múltiple, reportó:
· Biopsias:
- Peritoneo: positivas para la presencia de material amiloide.
- Médula ósea: positiva para presencia de material amiloide.¨ Inmunohistoquímica no concluyente para definir el origen del tipo de material amiloide.
¨ Presencia de 5% de células plasmáticas.- Hígado: negativo para presencia de material amiloide.
Confirmó el diagnóstico de amiloidosis primaria con compromiso cardiaco y estuvo de acuerdo con el manejo con ciclofosfamida, talidomida y prednisolona. Adicionalmente, reconoció que el paciente no es candidato para transplante de médula ósea.
Introducción
Amylum y amylon son palabras que provienen del Griego y el Latín y que hacen referencia al material amilaceous de las plantas, descrito por primera vez en 1838 por el botánico alemán Matthias Schleiden (1).
La amiloidosis cardiaca es una forma de manifestación de una serie de patologías sistémicas (2, 3). Es una entidad poco conocida y muchas veces sub-diagnosticada, en la cual, aun contando con el diagnóstico de compromiso cardiaco, será necesario aclarar su origen debido a las múltiples causas e implicaciones en su manejo y pronóstico.
La característica común de este grupo de enfermedades es el depósito extracelular de un material proteínico que cuando se tiñe con rojo Congo, demuestra birrefringencia en virtud luz polarizada color verde manzana. Evaluados con microscopio electrónico, el amiloide es visto como material fibrilar tipo hoja Beta. Son fibras no ramificadas con un diámetro de 7,5 a 10 nm de material proteico que se acumula por inadecuado plegamiento (4-6).
Las consecuencias del compromiso cardiaco pueden ser el motivo de consulta o por el contrario ser resultado de la búsqueda de compromiso de otros órganos involucrados. La presencia de amiloidosis cardiaca y su relativa predominancia, varía de acuerdo con el tipo de amiloidosis; así, la senil, la sistémica y algunas formas de amiloidosis asociadas a defectos en la transtirretín afectan de manera invariable el corazón, en cambio otras tienen un comportamiento variable como es el caso de la amiloidosis derivada de precursores de cadenas ligeras que puede pasar inadvertida o desarrollar un cuadro severo y maligno de falla cardiaca. Los casos de amiloidosis secundaria casi nunca comprometen el corazón (7). En la tabla 1 se observa la composición de las fibras de amiloidosis.
La amiloidosis cardiaca se caracteriza por la infiltración de amiloide extracelular en todo el corazón. Los depósitos de amiloide comprometen todas las estructuras cardiacas. El proceso infiltrante conduce a un engrosamiento de la pared biventricular sin generar dilatación ventricular, situación que altera los mecanismos de relajación diastólica y se asocia con el consiguiente aumento de la presión en las aurículas y como consecuencia con su dilatación.
El compromiso cardiaco en muy frecuente y puede coexistir con la disfunción de otros órganos; se entiende entonces a la amiloidosis cardiaca como una entidad con compromiso multisistémico.
AL amiloidosis
La forma más común de amiloidosis es la que se asocia a la discrasia de las células plasmáticas. El amiloide es generado a partir de un clon celular que produce de forma anormal cadenas ligeras de inmunoglobulinas y es reconocida como AL amiloidosis. La discrasia sanguínea que más se relaciona con ésta es el mieloma múltiple; ambas patologías tienen muchas cosas en común.
Sólo un pequeño grupo de estos pacientes desarrolla ambas patologías y la mayoría nunca desarrollarán mieloma múltiple. La AL amiloidosis se considera como una entidad poco común, pero tiene una frecuencia de aparición similar a las más conocidas enfermedades como la de Hodgkin o la leucemia mielocítica crónica (8).
El corazón se ve afectado en más de 50% de los casos de AL amiloidosis y la falla cardiaca es la forma de presentación en más de 50% de ellos. Una vez aparecen los síntomas de falla cardiaca, la supervivencia promedio es menor a seis meses (9, 10).
Manifestaciones clínicas
La insuficiencia cardiaca secundaria a AL amiloidosis se presenta en la mayoría de los casos con signos y síntomas de una enfermedad rápidamente progresiva. La disnea es un síntoma muy común y casi siempre se asocia con evidencia de la elevación de las presiones de llenado en las cavidades del lado derecho. El edema periférico suele ser progresivo y de muy difícil manejo farmacológico. La pérdida de peso puede representar el efecto de la enfermedad sistémica o ser la manifestación de la caquexia cardiaca. La presencia de disconfort torácico, no siendo típico de angina, en la mayoría de los casos es un síntoma secundario a la falla cardiaca (11). Algunas veces pueden presentarse con cuadros típicos de angina por compromiso de la microcirculación cardiaca. Los estudios no invasivos tienen altas tasas de falsos positivos aun en presencia de marcadores enzimáticos de necrosis miocárdica, lo cual se explica por el compromiso micro vascular (12). Presumiblemente, la elevación de la troponina implica necrosis miocárdica y se ha relacionado como un factor de mal pronóstico. El compromiso de los pequeños vasos por material amiloide puede producirse en ausencia de engrosamiento de la pared ventricular en el ecocardiograma.
La muerte súbita es común en AL amiloidosis; se ha demostrado que luego de un seguimiento a pacientes con falla cardiaca terminal secundaria a AL amiloidosis la causa de la muerte a menudo se debe a disociación electromecánica en lugar de arritmia ventricular (13). La taquicardia ventricular sostenida o la reanimación posterior a un episodio de fibrilación ventricular, es una forma rara de presentación que ocurre en pacientes con insuficiencia cardiaca menos severa, presumiblemente porque los pacientes con enfermedad más avanzada no sobreviven a un episodio inicial (14).
Menos de 5% de los pacientes con AL amiloidosis y compromiso del corazón tienen enfermedad cardiaca aislada. Las quejas que emiten como origen los síntomas no cardiacos, deben tenerse en cuenta porque su presencia es la idea de la naturaleza sistémica de la enfermedad (7). El paciente debe ser cuidadosamente interrogado acerca de la presencia de mareos y episodios de síncope con énfasis en la posición en la cual se desarrollan de esos síntomas ya que hay varios posibles mecanismos del síncope en la amiloidosis.
Durante la evaluación cardiovascular de un paciente con falla cardiaca secundaria a amiloidosis, en la mayoría de los casos se demuestra ritmo sinusal. La fibrilación auricular es la taquiarrítmia más frecuente y ocurre en 10% a 15% de los pacientes. Cuando sucede, el riesgo trombo-embólico aumenta notoriamente. El pulso venoso yugular puede ser muy evidente y a diferencia de la pericarditis constrictiva, el signo de Kussmaul está ausente (15, 16). El punto de máximo impulso en la mayoría de los casos no es palpable. Muy raras veces es posible demostrar su presencia aun en estadios terminales de S3 o S4. Las cifras de presión arterial son bajas en la mayoría de los casos, en ausencia de hipotensión ortostática (17). Es frecuente encontrar cuadros de hipotensión ortostática secundarios a disfunción autonómica como resultado de la neuropatía que pueden padecer los pacientes que cursan con cuadros de amiloidosis sistémica. La hipertensión arterial es inusual y se describen casos de resolución espontánea de la misma (18).
Los derrames pleurales bilaterales son comunes en gran parte de los casos y en la mayoría son secundarios al cuadro de falla cardiaca, pero puede coexistir infiltración pleural por amiloide, situación responsable de la pobre respuesta al tratamiento farmacológico y su rápida aparición posterior a su drenaje (19).
La hepatomegalia está presente bajo ciertas condiciones clínicas; la primera por congestión en pacientes con falla cardiaca, en la cual se encontrará el hígado levemente aumentado, blando y doloroso. La segunda por infiltración del órgano y se encontrará mayor crecimiento, varios centímetros por debajo del reborde costal, atravesando la línea media y de consistencia pétrea.
El edema periférico es progresivo y severo; en la mayoría de los casos no guarda relación con la severidad del síndrome de falla cardiaca. En estos pacientes es necesario descartar la presencia concomitante de síndrome nefrótico. La disfunción autonómica también es responsable de neuropatía sensitiva y los pacientes consultan por síntomas como parestesias y dolor neuropático (10).
Durante el análisis del electrocardiograma el hallazgo más frecuente es el bajo voltaje generalizado y en la mayoría de los casos se asocia a desviación importante del eje. Se ha descrito la presencia de criterios de hipertrofia del ventrículo izquierdo demostrado en precordiales, pero en estos casos se asocia a una entidad secundaria como hipertensión arterial (9, 20). Muy raras veces se describen bloqueos de rama del haz de His, siendo más usual el compromiso de la rama derecha.
Los hallazgos ecocardiográficos de la amiloidosis cardiaca avanzada, son múltiples. Se reconocen la hipertrofia concéntrica, el engrosamiento del ventrículo izquierdo, y del ventrículo derecho, la prominencia valvular y la infiltración del séptum atrial. La textura del miocardio es anormal y se describe como «granular» (21, 22). Con los avances disponibles en las técnicas Doppler se ha cambiado del patrón granular al reconocimiento de mayor ecogenicidad del miocardio y del tejido valvular.
Son hallazgos de enfermedad terminal la presencia de un patrón restrictivo por ecocardiografía Doppler y el aumento de ecogenicidad asociados al engrosamiento biventricular valvular. Las características del Doppler dependen de la etapa de la enfermedad, con base en una serie de estudios que demuestran una progresión de la disfunción diastólica en cuanto avanza la infiltración del miocardio (23).
La ecocardiografía Doppler también es útil en amiloidosis cardiaca. En estadios avanzados de la enfermedad se evidencia un patrón de flujo restrictivo que se caracteriza por un tiempo corto de desaceleración de la onda E y una onda A de baja velocidad, asociados con anomalías en el flujo venoso pulmonar (25). La disminución de la onda A transmitral en AL amiloidosis, se relaciona no sólo con la fase tardía de la fisiopatología restrictiva, sino también con el patrón de infiltración auricular amiloide, lo que da lugar a una disfunción intrínseca de la aurícula. Mediante imágenes de Doppler pulsado de tejidos, puede obtenerse información adicional sobre la función cardiaca en AL amiloidosis, que puede demostrar la presencia de disfunción diastólica con más exactitud que el flujo transmitral y pulmonar y puede proporcionar pruebas del deterioro del acortamiento longitudinal sistólico antes de que se altere la fracción de eyección (26, 27).
La función del ventrículo izquierdo está preservada hasta las etapas finales de la enfermedad, debido a que el ventrículo izquierdo no se dilata. La reducción de la fracción de eyección se asocia con disminución sustancial en el volumen de eyección. Aproximadamente en 5% de los pacientes con amiloidosis cardiaca, la infiltración del ventrículo izquierdo puede simular los hallazgos de la cardiomiopatía hipertrófica en el ecocardiograma (24). A diferencia de la verdadera cardiomiopatía hipertrófica, raras veces se ve hipertrofia ventricular en las derivaciones de las extremidades del electrocardiograma y el movimiento anterior sistólico de la válvula mitral es poco frecuente, aunque se puede observar el movimiento anterior de las cuerdas tendinosas y éstas se pueden acompañar de un soplo del tracto de salida del ventrículo izquierdo.
La asociación con hipotensión postural es común y la disminución de la postcarga cuenta para que la fracción de eyección se mantenga normal o incluso aumentada.
La resonancia magnética cardiaca es una de las ayudas imaginológicas más promisorias para el diagnóstico de la amiloidosis cardiaca, más aún en condiciones donde la ecocardiografía no ha sido diagnóstica. Las descripciones demuestran la presencia de un patrón inusual caracterizado por un realce tardío del Gadolinio en forma global que compromete todo el subendocardio (28).
Cateterismo cardiaco
Los estudios no invasivos son lo suficientemente eficientes para sugerir el diagnóstico de amiloidosis; por esto, la única indicación para realizar un procedimiento invasivo será para obtener una biopsia endomiocárdica que permita confirmar el diagnóstico histológico (29, 30).
Diagnóstico histológico
El diagnóstico de amiloidosis se basa en hacer evidente la presencia de birrefringencia verde manzana en el tejido al ser observada con un microscopio de luz polarizada luego de teñir con rojo Congo. Si los hallazgos ecocardiográficos son altamente sugestivos de amiloidosis, será posible obtener la muestra histológica de otros tejidos con procedimientos menos invasivos. Si no es posible obtener el diagnóstico tras la búsqueda en otros tejidos, será necesario realizar una biopsia endomiocárdica que en manos entrenadas se considera un procedimiento relativamente seguro; es virtualmente 100% sensible ya que la infiltración amiloide del corazón es uniforme (31, 32).
Una vez confirmado el diagnóstico histológico es fundamental descartar la presencia de una posible patología hematológica. Se deben realizar pruebas tipo inmunofijación en suero y orina más que electroforesis, porque la cantidad de paraproteína en éstas puede ser muy pequeña dado que la inmunofijación es una técnica mucho más sensible (33, 34). Está claro que el análisis del suero ofrece mayor precisión diagnóstica. En la AL amiloidosis están elevados los niveles de cadenas lambda o (menos comúnmente) kappa libres.
La relación de los niveles de las cadenas ligeras kappa y lambda libres deben evaluarse ya que a pesar de tener una excreción renal, condiciones como la insuficiencia renal eleva los niveles de las mismas sin cambiar la relación. Una proporción kappa a lambda mayor de 0,26, sugiere la presencia de una población clonal de células plasmáticas que produce cadenas ligeras lambda, mientras que una proporción mayor a 1,65 sugiere la producción clonal de cadenas ligeras kappa (35, 36).
Mediante el uso de las técnicas antes descritas es posible identificar hasta 99% de los pacientes que padecen esta enfermedad. El estudio de la médula ósea es parte fundamental del diagnóstico de esta patología ya que permite determinar la presencia de mieloma múltiple, clones responsables de la síntesis anormal de cadenas ligeras y de otras discrasias sanguíneas.
Es importante aclarar, como hallazgo normal, la presencia de una banda monoclonal en el estudio de immunofijación del suero en 5% a 10% de los pacientes mayores de 70 años de edad («gammapatía monoclonal de significado incierto») (37). La medición de cadenas ligeras libres en el suero es el estudio de elección en este tipo de casos; en otros de mayor complejidad se requieren estudios más sofisticados (38, 39).
Tratamiento
El tratamiento de la insuficiencia cardiaca se basa en los inhibidores de la enzima convertidora de angiotensina (no siempre es posible su uso debido a la baja presión arterial), los diuréticos y los beta-bloqueadores. El implante de un cardiodesfibrilador para el tratamiento de la arritmia maligna es controvertido y hasta ahora no existe suficiente evidencia clínica que avale este tipo de conducta. Los antagonistas del calcio y la digoxina están contraindicados en la AL amiloidosis, pero no hay ensayos clínicos que apoyen esta declaración relativamente antigua. Sólo existe un caso que informa sobre el transplante de corazón en pacientes con AL amiloidosis cardiaca. Los pacientes con enfermedad sistémica de células plasmáticas persistente, por lo general tienen un mal pronóstico después del transplante de corazón a causa de la recurrencia del material amiloide en los tejidos injertados. La posibilidad de altas dosis de melfalán y el transplante autólogo de células madre para erradicar el clon de células plasmáticas después de un transplante de corazón exitoso, debe investigarse en ensayos clínicos, pero los informes preliminares indican un posible beneficio de este enfoque (40-42).
El pilar del tratamiento de la insuficiencia cardiaca en la AL amiloidosis son los diuréticos, pero se ha visto necesario el uso de dosis más altas de lo previsto para lograr el mismo efecto clínico, debido a los niveles de albúmina más bajos como resultado de un síndrome nefrótico asociado. El grado de absorción por vía oral puede estar comprometido, situación por la cual se prefiere la administración endovenosa en estados congestivos. Los derrames pleurales pueden ser grandes y resistentes al tratamiento médico, lo cual puede indicar la presencia de amiloide pleural. Se pueden requerir drenajes recurrentes y en ocasiones pleurodesis.
Las terapias de elección para el manejo de falla cardiaca (inhibidores de la enzima convertidora de angiotensina y antagonistas de los receptores de angiotensina 2) pueden ser poco tolerados por hipotensión severa secundaria a la potenciación de la disfunción autonómica que presentan la mayoría de estos pacientes. Si se intenta iniciar este tipo de medicamentos, se deberá tener extrema precaución, idealmente con una muy baja dosis de captopril, elegido por su relativa corta vida media. No hay datos sobre la eficacia del beta-bloqueador sobre la supervivencia en la amiloidosis, pero su uso puede ser limitado a causa de la insuficiencia cardiaca refractaria o enfermedades relacionadas con la hipotensión grave. Los antagonistas del calcio están contraindicados por su efecto inotrópico negativo y la posible hipotensión asociada (43).
No hay datos publicados sobre el uso de fármacos del tipo inotrópicos o vasodilatadores en pacientes con insuficiencia cardiaca severa como consecuencia de amiloidosis. Sin embargo, podría ser posible el uso de dopamina a dosis bajas (1 a 3 mg/kg/min) como un complemento útil para el tratamiento de estados de anasarca, siempre y cuando la función renal sea adecuada. La digoxina puede provocar mayor riesgo de toxicidad por la alta afinidad de la molécula y el material amiloide (44). Sin embargo, cuando la fibrilación auricular con respuesta ventricular rápida está presente, la digoxina (administrada con precaución) puede ser por lo general segura y exitosa.
La presencia de fibrilación auricular en la AL amiloidosis, es una fuerte indicación para la anticoagulación con warfarina debido a la elevadísima tasa de eventos tromboembólicos. En la amiloidosis cardiaca avanzada, la aurícula está infiltrada, lo cual ocasiona su disfunción y favorece lo formación de trombos incluso en ritmo sinusal. Por lo tanto, es prudente anticoagular a todos los pacientes con AL amiloidosis, a pesar de estar en ritmo sinusal.
El tratamiento definitivo de la AL amiloidosis es la terapia anti-células plasmáticas destinada a poner fin a la producción de la paraproteína responsable de la formación del material amiloide (45). Existe una serie de regímenes de quimioterapia, pero el esquema con la mayor tasa de éxito parece ser el que incluye el uso de melfalán endovenoso, con una respuesta hematológica completa cercana a 40% de los pacientes que sobreviven a un año después de la quimioterapia (46). Lamentablemente, la naturaleza progresiva de la enfermedad cardiaca al momento del diagnóstico o bien no ser apto para las dosis altas de la quimioterapia con el posterior transplante autólogo de células madre, ubica a estos pacientes en un riesgo de mortalidad peri-tratamiento tan alto como de 30%. Los criterios precisos para definir un subgrupo de pacientes con AL amiloidosis que tienen baja la mortalidad frente al tratamiento en pacientes con AL amiloidosis y compromiso cardiaco, han sido difíciles de definir, pero la ausencia de insuficiencia cardiaca, una fracción de eyección normal y la carencia de líquido pleural parecen augurar un mejor pronóstico.
En contraste, encontrar un marcado engrosamiento de los ventrículos, elevación marcada del péptido natriurético cerebral o elevación de la troponina presagia un mal resultado. Los pacientes más jóvenes y las personas sin compromiso significativo de otros órganos o sistemas, también tienen mejores probabilidades de sobrevivir a la quimioterapia, pero en forma inesperada se han reportado arritmias, episodios de disociación electromecánica e incluso empeoramiento de la insuficiencia cardiaca en un grupo considerado de bajo riesgo (47). Tener una fracción de eyección de 40% generalmente se considera una contraindicación absoluta para recibir altas dosis de quimioterapia en un paciente con amiloidosis cardiaca, sobre todo porque la mayoría de estos pacientes tienen una clase funcional III de New York y una reserva cardiaca muy disminuida.
Sin embargo y a pesar del altísimo riesgo de muerte asociado con el uso de quimioterapia intensiva en los pacientes con AL amiloidosis y el compromiso cardiaco, se debe considerar en pacientes seleccionados ya que los supervivientes a menudo tienen una mejoría clínica en la falla cardiaca a pesar de no presentar cambios en su apariencia ecocardiográfica. La mejoría del síndrome de falla cardiaca puede obedecer a la abolición en la producción de nuevas cadenas ligeras, que son tóxicas para las células del miocardio, lo que sugiere que la AL amiloidosis no es sólo una miocardiopatía infiltrativa sino más bien un desorden infiltrante cardio-tóxico (48, 49). Los esquemas recomendados para el manejo de la AL amiloidosis con compromiso cardiaco, fueron recientemente revisados y se anotan en la tabla 2.
Para los pacientes que no toleran dosis altas de melfalán por vía intravenosa, existen datos preliminares provenientes del grupo de Amilodosis del Reino Unido que sugieren una modificación del régimen del medicamento en mención, a dosis mensuales que pueden ser mejor soportadas con una tasa de respuesta similar. No obstante, a la fecha no existe un estudio comparativo de estas dos terapias. El «régimen estándar» de melfalán y prednisona dada como «pulsos» de dosis de tres a cinco días cada seis semanas, parece generar poco beneficio en pacientes con amiloidosis cardiaca, probablemente porque se requieren varios meses para ver un efecto (50). Además, el régimen de esteroides puede agravar la insuficiencia cardiaca congestiva. Recientemente, se ha utilizado una dosis baja de un régimen «continuo» de melfalán en pacientes con amiloidosis cardiaca severa en quienes se evidencia respuesta hematológica en siete de trece individuos estudiados. Infortunadamente, la enfermedad cardiaca a menudo es muy grave al inicio del tratamiento para determinar si un régimen de este tipo tenga un impacto en la supervivencia a largo plazo. Regímenes que incluyen el uso de dosis altas de dexametasona, como vincristina, adriamicina y dexametasona por lo general no son tolerados en los cuadros de amiloidosis cardiaca (51), en primera instancia porque la adriamicina, aunque sea empleada en dosis relativamente pequeñas, puede producir toxicidad cardiaca y la dexametasona, por su parte, puede empeorar la insuficiencia cardiaca.
En casos muy seleccionados, puede considerarse el transplante cardiaco. La experiencia con este procedimiento en la AL amiloidosis, sugirió que a corto y mediano plazo la mortalidad no fue diferente a la de otros transplantes (52), pero un informe posterior de una pequeña serie de pacientes tratados en varios centros de transplante, demostró una aparente mayor mortalidad a largo plazo de la esperada, por lo general debido a la progresión de la enfermedad cardiaca o en los demás órganos comprometidos (53, 54). Como resultado de estas observaciones, muchos centros de transplante lo consideran una contraindicación. Sin embargo, con la llegada de nuevos esquemas de quimioterapia y el avance en el transplante de células madre, será posible el transplante de corazón para luego llevar a cabo la quimioterapia durante seis a doce meses y así poder suprimir la producción de material amiloide. Los posibles candidatos para este tipo de procedimiento combinado, son poco frecuentes ya que el compromiso de otros órganos es una contraindicación y la enfermedad clínicamente limitada al corazón sólo se da en menos de 5% de los casos. Sin embargo, se ha tenido éxito en el tratamiento de un número de pacientes con este enfoque combinado, y varios de ellos obtuvieron una remisión de la enfermedad a largo plazo, sin evidencia de recidiva después de tres a cinco años de seguimiento.
Bibliografía
1. Hassan W, Sergani HA, Mourad W, et al. Amyloid heart disease: new frontiers and insights in pathophysiology, diagnosis, and management. Tex Heart Inst J 2005; 32: 178-84. [ Links ]
2. Marín JE, Duque M, Medina LE, et al. Cardiopatía amiloide. Rev Col Cardiol 2005; 11: 389-396. [ Links ]
3. Falk Rodney H. Diagnosis and management of the cardiac amyloidoses. Circulation 2005; 112: 2047-2060. [ Links ]
4. Sipe JD, Cohen AS. History of the amyloid fibril. J Struct Biol 2000; 130: 88-98. [ Links ]
5. Selkoe DJ. Folding proteins in fatal ways. Nature 2003; 426: 900-904. [ Links ]
6. Merlini G, Westermark P. The systemic amyloidoses: clearer understanding of the molecular mechanisms offers hope for more effective therapies. J Intern Med 2004; 255: 159-178. [ Links ]
7. Dubrey SW, Cha K, Simms RW, Skinner M, Falk RH. Electrocardiography and Doppler echocardiography in secondary (AA) amyloidosis. Am J Cardiol 1996; 77: 313-315. [ Links ]
8. Gertz MA, Lacy MQ, Dispenzieri A. Amyloidosis hematology. Oncol Clin North Am 1999; 13: 1211-1220. [ Links ]
9. Dubrey SW, Cha K, Anderson J, Chamarthi B, Reisinger J, Skinner M, Falk RH. The clinical features of immunoglobulin light-chain (AL) amyloidosis with heart involvement. QJM 1998; 91: 141-157. [ Links ]
10. Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol 1995; 32: 45-59. [ Links ]
11. Mueller PS, Edwards WD, Gertz MA. Symptomatic ischemic heart disease resulting from obstructive intramural coronary amyloidosis. Am J Med 2000; 109: 181-188. [ Links ]
12. Zabernigg A, Schranzhofer R, Kreczy A, Gattringer K. Continuously elevated cardiac troponin I in two patients with multiple myeloma and fatal cardiac amyloidosis. Ann Oncol 2003; 14: 1791. [ Links ]
13. Falk RH, Rubinow A, Cohen AS. Cardiac arrhythmias in systemic amyloidosis: correlation with echocardiographic abnormalities. J Am Coll Cardiol 1984; 3: 107-113. [ Links ]
14. Chamarthi B, Dubrey SW, Cha K, Skinner M, Falk RH. Features and prognosis of exertional syncope in light-chain associated AL cardiac amyloidosis. Am J Cardiol 1997; 80: 1242-1245. [ Links ]
15. Klein AL, Cohen GI. Doppler echocardiographic assessment of constrictive pericarditis, cardiac amyloidosis, and cardiac tamponade. Cleveland Clin J Med 1992; 59: 278-290. [ Links ]
16. Tyberg TI, Goodyer AV, Hurst VWD, Alexander J, Langou RA. Left ventricular filling in differentiating restrictive amyloid cardiomyopathy and constrictive pericarditis. Am J Cardiol 1981; 47: 791-796. [ Links ]
17. Murphy L, Falk RH. Left atrial kinetic energy in AL amyloidosis: can it detect early dysfunction? Am J Cardiol 2000; 86: 244-246. [ Links ]
18. Bernardi L, Passino C, Porta C, Anesi E, Palladini G, Merlini G. Widespread cardiovascular autonomic dysfunction in primary amyloidosis: does spontaneous hyperventilation have a compensatory role against postural hypotension? Heart 2002; 88: 615-621. [ Links ]
19. Berk JL, Keane J, Seldin DC, Sanchorawala V, Koyama J, Dember LM, Falk RH. Persistent pleural effusions in primary systemic amyloidosis: etiology and prognosis. Chest 2003; 124: 969-977. [ Links ]
20. Murtagh B, Hammill SC, Gertz MA, Kyle RA, Tajik AJ, Grogan M. Electrocardiographic findings in primary systemic amyloidosis and biopsy-proven cardiac involvement. Am J Cardiol 2005; 95: 535-537. [ Links ]
21. Siqueira-Filho AG, Cunha CL, Tajik AJ, Seward JB, Schattenberg TT, Giuliani ER. M-mode and two-dimensional echocardiographic features in cardiac amyloidosis. Circulation 1981; 63: 188-196. [ Links ]
22. Child JS, Levisman JA, Abbasi AS, MacAlpin RN. Echocardiographic manifestations of infiltrative cardiomyopathy: a report of seven cases due to amyloid. Chest 1976; 70: 726-731. [ Links ]
23. Klein AL, Hatle LK, Taliercio CP, Taylor CL, Kyle RA, Bailey KR, Seward JB, Tajik AJ. Serial Doppler echocardiographic follow-up of left ventricular diastolic function in cardiac amyloidosis. J Am Coll Cardiol 1990; 16: 1135-1141. [ Links ]
24. Sedlis SP, Saffitz JE, Schwob VS, Jaffe AS. Cardiac amyloidosis simulating hypertrophic cardiomyopathy. Am J Cardiol 1984; 53: 969-970. [ Links ]
25. Klein AL, Hatle LK, Burstow DJ, Seward JB, Kyle RA, Bailey KR, Luscher TF, Gertz MA, Tajik AJ. Doppler characterization of left ventricular diastolic function in cardiac amyloidosis. J Am Coll Cardiol 1989; 13: 1017-1026. [ Links ]
26. Abdalla I, Murray RD, Lee JC, Stewart WJ, Tajik AJ, Klein AL. Duration of pulmonary venous atrial reversal flow velocity and mitral inflow a wave: new measure of severity of cardiac amyloidosis. J Am Soc Echocardiogr 1998; 11: 1125-1133. [ Links ]
27. Koyama J, Davidoff R, Falk RH. Longitudinal myocardial velocity gradient derived from pulsed Doppler tissue imaging in AL amyloidosis: a sensitive indicator of systolic and diastolic dysfunction. J Am Soc Echocardiogr 2004; 17: 36-44. [ Links ]
28. Kwong RY, Falk RH. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation 2005; 111: 122-124. [ Links ]
29. Swanton RH, Brooksby IA, Davies MJ, Coltart DJ, Jenkins BS, Webb- Peploe MM. Systolic and diastolic ventricular function in cardiac amyloidosis: studies in six cases diagnosed with endomyocardial biopsy. Am J Cardiol 1977; 39: 658-664. [ Links ]
30. Robbins MA, Pizzarello RA, Stechel RP, Chiaramida SA, Gulotta SJ. Resting and exercise hemodynamics in constrictive pericarditis and a case of cardiac amyloidosis mimicking constriction. Cathet Cardiovasc Diagn 1983; 9: 463-471. [ Links ]
31. Pomerance A, Slavin G, McWatt J. Experience with the sodium sulphate-Alcian blue stain for amyloid in cardiac pathology. J Clin Pathol 1976; 29: 22-26. [ Links ]
32. Libbey CA, Skinner M, Cohen AS. Use of abdominal fat tissue aspirate in the diagnosis of systemic amyloidosis. Arch Intern Med 1983; 143: 1549-1552. [ Links ]
33. Abraham RS, Katzmann JA, Clark RJ, Bradwell AR, Kyle RA, Gertz MA. Quantitative analysis of serum free light chains: a new marker for the diagnostic evaluation of primary systemic amyloidosis. Am J Clin Pathol 2003; 119: 274-278. [ Links ]
34. Katzmann JA, Clark RJ, Roshini S, Abraham RS, Bryant S, Lymp JF, et al. Serum reference intervals and diagnostic ranges for free and free immunoglobulin light chains: relative sensitivity for detection of monoclonal light chains. Clin Chem 2002; 48: 1437-1444. [ Links ]
35. Katzmann JA, Abraham RS, Dispenzieri A, Lust JA, Kyle RA. Diagnostic performance of quantitative kappa and lambda free light chain assays in clinical practice. Clin Chem 2005; 51: 878-881. [ Links ]
36. Swan N, Skinner M, O'Hara CJ. Bone marrow core biopsy specimens in AL (primary) amyloidosis: a morphologic and immunohistochemical study of 100 cases. Am J Clin Pathol 2003; 120: 610-616. [ Links ]
37. Kyle RA, Therneau TM, Rajkumar SV, Larson DR, Plevak MF, Melton LJ. Long-term follow-up of 241 patients with monoclonal gammopathy of undetermined significance: the original Mayo Clinic series 25 years later. Mayo Clin Proc 2004; 79: 859-866. [ Links ]
38. O'Hara CJ, Falk RH. The diagnosis and typing of cardiac amyloidosis. Amyloid 2003; 10: 127-129. [ Links ]
39. Lachmann HJ, Booth DR, Booth SE, Bybee A, Gilbertson JA, Gillmore JD, et al. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med 2002; 346: 1786-1791. [ Links ]
40. Röcken C, Ernst J, Hund E, Michels H. et al. Interdisciplinary guidelines on the diagnosis of and therapy for systemic amyloidosis. Dtsch Med Wochenschr 2006; 131: S45-S66. [ Links ]
41. Alloni A, Pellegrini C, Ragni T, Goggi C, D'Armini AM, Rinaldi M, Vigano M. Heart transplantation in patients with amyloidosis: single-center experience. Transplant Proc 2004; 36: 643-644. [ Links ]
42. Mohty M, Albat B, Fegueux N, Rossi JF. Autologous peripheral blood stem cell transplantation following heart transplantation for primary systemic amyloidosis. Leuk Lymphoma 2001; 41: 221-223. [ Links ]
43. Gertz MA, Falk RH, Skinner M, Cohen AS, Kyle RA. Worsening of congestive heart failure in amyloid heart disease treated by calcium channel-blocking agents. Am J Cardiol 1985; 55: 1645. [ Links ]
44. Rubinow A, Skinner M, Cohen AS. Digoxin sensitivity in amyloid cardiomyopathy. Circulation 1981; 63: 1285-1288. [ Links ]
45. De Lorenzi E, Giorgetti S, Grossi S, Merlini G, Caccialanza G, Bellotti V. Pharmaceutical strategies against amyloidosis: old and new drugs in targeting a "protein misfolding disease". Curr Med Chem 2004; 11: 1065-1084. [ Links ]
46. Skinner M, Sanchorawala V, Seldin DC, Dember LM, Falk RH, Berk JL, et al. High-dose melphalan and autologous stem-cell transplantation in patients with AL amyloidosis: an 8-year study. Ann Intern Med 2004; 140: 85-93. [ Links ]
47. Falk RH, Reisinger J, Dubrey SW, Mendes LA, Sanchorawala V, Ekery D, et al. The effect of cardiac involvement on the outcome of intravenous melphalan therapy and autologous stem cell rescue for AL amyloidosis. In: Kyle RA, Gertz MA, eds. Amyloid and the Amyloidoses: VIIIIth International Symposium on Amyloidosis. Rochester, Minn: Parthenon; 1998. p. 181-183. [ Links ]
48. Liao R, Jain M, Teller P, Connors LH, Ngoy S, Skinner M, et al. Infusion of light chains from patients with cardiac amyloidosis causes diastolic dysfunction in isolated mouse hearts. Circulation 2001; 104: 1594-1597. [ Links ]
49. Brenner DA, Jain M, Pimentel DR, Wang B, Connors LH, Skinner M, et al. Human amyloidogenic light chains directly impair cardiomyocyte function through an increase in cellular oxidant stress. Circ Res 2004; 94: 1008-1010. [ Links ]
50. Skinner M, Anderson J, Simms R, Falk R, Wang M, Libbey C, et al. Treatment of 100 patients with primary amyloidosis: a randomized trial of melphalan, prednisone, and colchicine versus colchicine only. Am J Med 1996; 100: 290-298. [ Links ]
51. Wardley AM, Jayson GC, Goldsmith DJ, Venning MC, Ackrill P, Scarffe JH. The treatment of nephrotic syndrome caused by primary (light chain) amyloid with vincristine, doxorubicin and dexamethasone. Br J Cancer 1998; 78: 774-776. [ Links ]
52. Hosenpud JD, Uretsky BF, Griffith BP, O'Connell JB, Olivari MT, Valantine HA. Successful intermediate-term outcome for patients with cardiac amyloidosis undergoing heart transplantation: results of a multicenter survey. J Heart Transplant 1990; 9: 346-350. [ Links ]
53. Dubrey SW, Burke MM, Hawkins PN, Banner NR. Cardiac transplantation for amyloid heart disease: the United Kingdom experience. J Heart Lung Transplant 2004; 23: 1142-1153. [ Links ]
54. Hosenpud JD, DeMarco T, Frazier OH, Griffith BP, Uretsky BF, Menkis AH, et al. Progression of systemic disease and reduced long-term survival in patients with cardiac amyloidosis undergoing heart transplantation: follow-up results of a multicenter survey. Circulation 1991; 84: III-338-II-43. [ Links ]

