










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Colombiana de Cardiología
versión impresa ISSN 0120-5633
Rev. Colom. Cardiol. v.18 n.1 Bogota ene./feb. 2011
Arritmias potenciadas por isquemia sub-epicárdica en pared transmural heterogénea cardiaca: un estudio teórico de simulación
Arrhythmias potentiated by sub-epicardial ischemia in transmural heterogeneous cardiac wall: theoretical simulation study
(1) Instituto de Investigación Interuniversitario en Bioingeniería y Tecnología Orientada al Ser Humano, Universidad Politécnica de Valencia.Valencia, España.
(2) Laboratorio de Fisiología Celular e Inmunología, Universidad Tecnológica de Pereira. Pereira, Colombia.
(3) Grupo de Seguimiento al Corazón Vía Satélite, Ciudadela Salud Bogotá. Bogotá, Colombia.
Correspondencia: Dr. Óscar A. Henao Gallo, Celular: 310-504 28 17. Correo electrónico: oshegal@aaa.upv.esoscarhe@utp.edu.co.
Recibido: 30/09/2010. Aceptado: 09/11/2010.
La fibrilación ventricular, la isquemia miocárdica y la muerte súbita son fisiopatologías cardiacas inseparables. La influencia de la distribución de células del medio miocardio en la formación de arritmias en la pared heterogénea cardiaca en presencia de isquemia sub-epicárdica, no está del todo dilucidada. En este estudio se modela una porción plana de la pared transmural con diferentes configuraciones de células del medio miocardio que se adjuntan a las heterogeneidades bioquímicas presentes en isquemia sub-epicárdica para cuantificar su influencia en la formación de arritmias.
Se obtuvieron reentradas lobulares no sostenidas en torno de la lesión isquémica que interfieren con las células M, alterando la repolarización del tejido. La función de vulnerabilidad que cuantifica la prospección a reentradas es aproximada por una función logística, y su mayor expresión ocurre en el minuto 8,75 de isquemia modelada.
La heterogeneidad bioquímica y morfológica en el tejido virtual estudiado dan como resultado una arritmia por reentrada; su secuela en la vulnerabilidad del tejido aumenta a medida que crece la severidad de la hiperkalemia. Los electrogramas obtenidos muestran depresión TQ y elevación ST con una morfología de taquicardia ventricular polimórfica.
PALABRAS CLAVE: células M, electrogramas, función de vulnerabilidad, isquemia sub-epicárdica, modelo de Luo-Rudy, reentrada transmural, taquicardia polimórfica.
Ventricular fibrillation, myocardial ischemia and sudden cardiac death are inseparable cardiac pathophysiologies. The influence of the distribution of myocardial cells in the formation of arrhythmias in the heterogeneous cardiac wall in the presence of sub-epicardial ischemia is not entirely elucidated. This study models a flat portion of the transmural wall under different myocardial cell configurations attached to the biochemical heterogeneity present in sub-epicardial ischemia to quantify their influence on the development of arrhythmias.
Lobular non-sustained reentries were obtained around the ischemic lesion that interfere with M cells, altering the tissue repolarization. Vulnerability function that quantifies prospection to reentries is approximated by a logistic function, and its main expression occurs in 8.75 minutes of modeled ischemia.
The biochemical and morphological heterogeneity in the virtual tissue studied results in a reentrant arrhythmia; its sequel to the tissue vulnerability increases as the severity of hyperkalemia grows. Electrograms obtained show TQ depression and ST elevation with a morphology of polymorphic ventricular tachycardia.
KEY WORDS: M cells, electrograms, vulnerability function, sub-epicardial ischemia, Luo-Rudy model, transmural reentry, polymorphic tachycardia.
Introducción
Hasta finales de la década de los ochenta, se creía que los corazones sanos sólo tenían dos tipos de células que lo conformaban: el sistema de conducción His-Punkinje y las del miocardio. En 1988 Litovsky y Antzelevitch (1, 2) describieron por primera vez las diferencias esenciales entre células epicárdicas y endocárdicas para luego caracterizar, de manera unívoca, un conjunto de células del medio miocardio con unas características particulares en la fase de repolarización del potencial (3). Las células del medio miocardio (células M) se diferencian del epicardio y el endocardio en la expresión de densidad de canales asociados con la activación débil y lenta de la corriente de rectificación retardada de potasio (IKs) produciendo alargamiento en los potenciales de acción y la corriente transitoria de salida de potasio (Ito) produciendo la morfología de espiga y domo en epicardio y medio miocardio, además de una larga corriente tardía de sodio (INa_late) y del intercambiador sodio_calcio (INa_Ca) (4).
Los gradientes transmurales de repolarización se presentan por la aparición de una ranura mediada por (Ito) en epicardio pero no en endocardio, siendo estos los que expresan la onda Osborne (J) del electrocardiograma (5). Gradientes de voltaje desarrollados por diferencia temporal en la fase 2 y 3 del potencial en los tres tipos de célula, conllevan la formación de gradientes de voltaje de signo opuesto lateralmente concomitante con las células M siendo responsables en mayor parte de la inscripción de la onda T del electrocardiograma (6, 7). Estos gradientes son sensitivos de modulación por comunicación electrotónica entre los tres tipos de células, y aumento de [K+]o, lo cual conduce a alteraciones de la onda T del electrograma (8-10). Una reducción en la corriente de repolarización conlleva prolongamiento preferencial del potencial en células M, responsable de la prolongación del intervalo QT e incrementa la dispersión transmural de repolarización la cual delinea arritmias tipo torsades de pointes (11, 12).
La isquemia aguda afecta la incidencia de arritmias en enfermedad coronaria de diversas formas, haciendo imposible investigar su intrincada fisiopatología en modelos experimentales diseñados sólo para medir algunos de sus parámetros relevantes (13-15) (aumento de concentraciones iónicas intra y extracelular, falta de oxigenación e intercambio de gases y metabolitos (16-18). De igual manera, la isquemia aguda genera muchos cambios bioquímicos que influencian la señal eléctrica producida por los miocitos (ver Janse (19) , Carmeliet (20) para una revisión). Por lo anterior, el estudio de la isquemia aguda utilizando modelos matemáticos biofísicamente detallados que incluyen balance iónico, aumento de potasio extracelular (13), corriente de potasio modulada por ATP (21), heterogeneidad celular (22, 23) y anisotropía en la conducción del impulso (24) permite entender el papel preponderante de la isquemia en el desarrollo de arritmias fatales en la pared transmural (25).
En este trabajo se presenta un modelo detallado de isquemia aguda regional sub-epicárdica en un tejido virtual transmural bidimensional, en el cual se diseñan de manera realista diferentes arreglos conformacionales de células del medio miocardio.
La actividad eléctrica de las células es descrita por un modelo de potencial de acción detallado de Luo-Rudy (23, 26, 27) (versión 2000). El modelo se usó para estudiar la predisposición a generar los diferentes patrones de reentrada que son causados por alteraciones bioquímicas del tejido generando taquicardia ventricular tanto en pared transmural heterogénea como en epicardio sujeto a isquemia regional.
Métodos
Es interés del grupo de investigadores estudiar la influencia de la heterogeneidad de la pared cardiaca (funcional, lesionada por isquemia) y sus mecanismos iónicos como generadores de patrones reentrantes en isquemia sub-epicárdica aguda. Sus diferentes alteraciones bioquímicas: hipoxia, acidosis e hiperkalemia han sido modelados e incluidos en la descripción cinética de iones a través de la membrana desarrollada por Luo-Rudy (28). La estructura matemática básica de una célula cardiaca del modelo es (ver parte A de la figura 1):
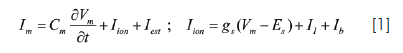
Donde Im es la corriente a través de la membrana, Cm es la capacitancia de la membrana celular, Vm es el potencial, Iion es la suma de las corrientes iónicas las bombas electrogénicas y los intercambiadores por unidad de área en la descripción de Luo-Rudy, y Iest es el estímulo de corriente aplicado. Cada corriente a través de un canal específico es descrita por el formalismo de Hodgkin-Huxley (29).
Formulación del monodominio
Si el tejido cardiaco se asume como un sincitio funcional (conglomerado de células que actúan como una unidad funcional) entonces se puede describir mediante la ecuación del cable desarrollada por Wiedmann (30). Una extensión de dicha ecuación para un tejido bidimensional espacialmente extendido se suscribe como:
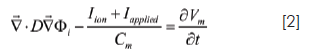
El tejido consiste en una malla de células interconectadas por medio de resistencias que representan las "gap junction". La cinética iónica se modela mediante Luo-Rudy. En todos los tejidos simulados (30 x 30 mm en epicardio y 30 x 15 mm en pared transmural), como el representado en la parte B de la figura 1, cada célula interna (sin fronteras) es modelada como un nodo con cuatro puntos adyacentes lineales conectados por resistencia (detalle en la parte A de la figura 1). Algunos estudios recientes (31, 32) dan soporte a dicho modelo, y por lo tanto cada conexión longitudinal y transversal puede ser programada de manera individual. La naturaleza microscópica de la estructura del ventrículo es ignorada y así el tejido es tratado como un sistema continuo modelado por una ecuación de reacción-difusión:
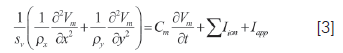
Donde Sv es el cociente entre superficie y volumen celular y rx y ry son las resistividades en la dirección transversal y longitudinal respectivamente. Por propósitos computacionales, el tejido fue discretizado en porciones de 100 x 100 mm. La ecuación [3] fue resuelta utilizando el método numérico de operador partido. Los valores de resistividades celulares fueron escogidos de tal forma que la velocidad de conducción en dirección longitudinal fue de 50 cm-1 y en dirección trasversal fue de 13 cm-1 con un cociente anisotrópico en la velocidad de aproximadamente 4:1 (33). Se utilizó protocolo de estimulación S1-S1 con un adecuado intervalo de acople (CI) para el pulso prematuro aplicado al tejido con isquemia regional. La amplitud del pulso de corriente aplicado es dos veces el umbral diastólico y su duración es de 2 ms.
Modelo de isquemia regional aguda
La parte B de la figura 1 muestra la estructura electrofisiológica del tejido modelado (correspondiente al minuto ocho después del comienzo de la isquemia), en el cual existen diferentes zonas que representan los cambios generados por la isquemia. El tamaño de estas zonas, así como los valores relevantes de los parámetros del modelo y sus gradientes espaciales, se tomaron de datos experimentales revisados por Coronel (34). Se identifican una zona central (CZ) lesionada, una zona en forma de aro correspondiente con la zona de borde (BZ) y una zona normal de tejido epicárdico (NZ) rodeando la zona de borde. Se mantiene la misma disposición geométrica para la lesión isquémica sub-epicárdica de la pared. Los valores de los parámetros que modelan la acidosis y la hiperkalemia en la lesión isquémica fueron descritos anteriormente por Ferrero (35). La corriente, la fracción de canales abiertos y la conductancia del canal considerados por el modelo se suscriben como:
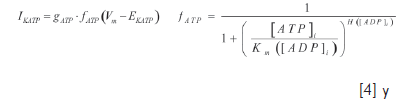
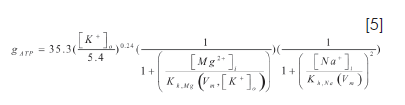
Los niveles de ATP y ADP proporcionan una fracción de canales dada por en la CZ lesionada (21). En la tabla 1 se describen los diferentes instantes de isquemia regional modelados en este estudio; se resalta que los canales de potasio dependientes de ATP son activados y mantenidos dentro de su valor normal para el modelo.
Modelo de heterogeneidad transmural
La heterogeneidad cardiaca es incluida en el modelo variando las conductancias de la corriente transitoria de salida Ito (22) y la componente lenta del rectificador inverso de potasio IKs (36) con una relación de 23:7:15 entre las células del epicardio: medio miocardio: endocardio respectivamente. Los valores de conductancia se alteraron con base en el trabajo de simulación de Clayton (37). Las diferentes configuraciones de células M se muestran en la parte C de la figura 1 y se basan en trabajos experimentales en porciones de tejido ventricular canino (38, 39) su distribución es en aglomerados circulares ocupando 35% de la pared, una banda que ocupa 55% de la pared, un diseño circular que alberga el 17% y un caso control donde no se modelan células del medio miocardio en la parte medial del tejido, siendo el epicardio 20% de la porción transmural (Henao describió detalles del modelo) (40).
Modelo de electrograma
La actividad eléctrica cardiaca involucra la forma del potencial de acción y su propagación a través del corazón. Esta actividad eléctrica determina las fuentes bioeléctricas. Los cambios en el volumen conductor son a menudo extra cardiacos y se reflejan en el electrograma (EG).
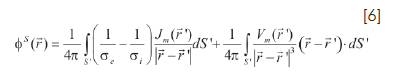
A partir del modelo de fuentes doble capa, Geselowitz mostró que el electrograma, que se calcula a partir del modelo de doble capa uniforme descrito por la ecuación [6], puede dar información diagnóstica sobre eventos acaecidos en el corazón (41). Se supone un electrodo de captación del electrograma a un milímetro del centro de la zona lesionada y allí se evalúa la señal de EG con base en una versión discreta de la ecuación [6].
Resultados
La figura 2 muestra el ciclo que conduce a eventos arrítmicos por reentrada y taquicardia polimórfica. Por un lado, fármacos o alteraciones en acople resistivo celular (42) pueden ocasionar una marcada dispersión en la duración del potencial que desarrollan los mecanismos de gatillo a través de la formación de despolarizaciones tempranas que pueden delinear los complejos prematuros que inician las características de la taquicardia ventricular, torsades de pointes en pacientes con síndrome de QT largo. La dispersión en la repolarización en la zona de la lesión genera una reentrada en figura de ocho en torno de la zona central, cuya manifestación electrocardiográfica es la aparición de potenciales dobles generados por el choque de los dos lóbulos reentrantes, potenciando la taquicardia ventricular polimórfica que se observa en el electrograma emn la parte media derecha de la figura 2. En el modelo de figura de ocho, el latido reentrante genera un frente de onda que circula en torno a una línea de bloqueo funcional, uniéndose en la parte distal del bloqueo. El singular arco de bloqueo se divide en dos partes y la activación reentrante continúa como dos frentes de onda circulantes que viajan a favor y en contra del movimiento de las manecillas del reloj en una configuración oblonga en ocho horizontal.
La figura 3 muestra el patrón reentrante en cuatro instantes predeterminados, para los cuatro minutos analizados en este estudio y para cada una de las configuraciones estudiadas. En la parte inferior de la figura se muestran los instantes prominentes, la propagación del estímulo básico (100 ms), la formación de la reentrada lobular después de aplicar el segundo estímulo prematuro (320 ms), el retraso y la dispersión de repolarización en la zona isquémica (460 ms) y el avance y la terminación autónoma del estímulo en la parte distal (590 ms). En el primer cuadro se nota la propagación del estímulo a través de la pared y resalta las diferentes configuraciones de células del medio miocardio. La formación lobular tiene igual configuración en todos los instantes; sólo se percibe una leve alteración en su proceso de repolarización al llegar a la zona central de la lesión en aquellas configuraciones que interfieren con ella. En el tercer cuadro se muestra una de las alteraciones más dramáticas; nótese el avance del frente de onda en las dos primeras configuraciones y la drástica disminución de este avance por alteraciones en la repolarización en la zona central. En las dos últimas configuraciones, el frente aun no ha empezado a excitar en dirección contraria la parte superior del tejido virtual. En el último cuadro, en la parte superior, se observa la detención del frente reentrante en la zona distal de la lesión, sin embargo a medida que avanza la severidad de la isquemia, en la escala vertical, el frente cada vez más excita menos tejido llegando a visualizarse la segunda reentrada lobular en la última configuración, debido al extenso retardo en la propagación del frente en la zona lesionada.
La figura 4 muestra los respectivos electrogramas de las simulaciones de la figura anterior, el primer pulso aplicado al tejido presenta una deflexión TQ (aproximadamente de 10 mV) debido a la severidad de la hiperkalemia en la zona de la lesión (Tabla 1), aparece un complejo QRS de 90 ms de duración, con una pequeña deformación de la onda S en las dos últimas configuraciones. Luego una elevación del segmento ST en todos los electrogramas y una amplia y deformada onda T cuyo retraso temporal va en aumento. Cuando se aplica el segundo estímulo al tejido, este interrumpe el final de la onda T del pulso anterior y hay una nueva despolarización de la zona isquémica cuya amplitud y duración está alterada. El electrograma de la parte A presenta un segundo pulso puntiagudo y cuasi simétrico cuya amplitud y duración son mayores que la del pulso de acondicionamiento, alterándose por completo la excitación del tejido y su respectiva repolarización. En los electrogramas de la parte B y C aparece el segundo pulso con dos pequeños y retardados picos en su onda R (de mayor tamaño en electrograma de la configuración de un círculo, respecto de la configuración control) y una profunda y retardada onda S que da información sobre la profunda alteración de la repolarización del tejido; el retraso en dichas ondas informa sobre alteraciones en la velocidad de conducción del frente de onda reentrante dentro del tejido virtual. El electrograma de la parte D muestra una onda R retardada y disminuida que representa lo lento de la propagación del frente por la zona isquémica, y una onda S de mayor profundidad que la del caso anterior donde se hace más evidente el cambio en la repolarización del tejido. Finalmente, el tramo correspondiente a la formación de la onda T del frente reentrante, cada vez es menor, evidenciándose mayor cantidad de tejido entrando a fase de repolarización disminuyendo la propagación del frente de onda en la zona isquémica, hasta detenerlo de manera autónoma en la parte distal de la región.
La figura 5 muestra explícitamente las funciones de vulnerabilidad para los minutos de isquemia estudiados en este trabajo y para cada una de las configuraciones utilizadas. En la parte A se muestra una comparación de las funciones: vulnerabilidad en términos de distribución (eje X organizadas de mayor a menor tamaño de células del medio miocardio), minuto estudiado (eje Y cuya duración se incrementa de derecha a izquierda) e intensidad (alturas variables de las columnas), en la cual se evidencia cómo la función de vulnerabilidad aumenta al doble si la zona de células del medio miocardio interfiere con la zona de la lesión (a la izquierda de la parte B) y disminuye de forma monótona en las dos distribuciones donde las células del medio miocardio no están presentes en la zona de la lesión (a la derecha de la parte B). Es de notarse el pequeño incremento de la vulnerabilidad en los primeros instantes estudiados, convirtiéndose en el doble y casi el triple de este intervalo en los dos últimos instantes analizados. Esta disparidad entre los intervalos, hace suponer un comportamiento de dos estados en las funciones de vulnerabilidad; así, un primer estado ha de ser un intervalo pequeño de 5 ó 6 ms y un segundo estado donde la probabilidad a presentar reentradas presenta un valor triple respecto del primero. Un análisis estadístico de este comportamiento permite aproximar estas funciones de vulnerabilidad por una función logística con una media de 4,60 y una desviación de 9,71 cuyo parámetro de comparación es el logaritmo de verosimilitud, y su valor de -443,58.
Discusión
Rol de curvatura del frente de onda
La curvatura local a lo largo del frente de onda, es un concepto clave en la formación, mantenimiento y terminación de la reentrada obtenida. Los cambios en la velocidad de propagación son dependientes de la curvatura del frente. La curvatura del frente reentrante es más grande cerca de la singularidad y decrece a medida que se aleja. La curvatura es limitada por la habilidad de una onda convexa para excitar el tejido delante de ella. La noción de curvatura crítica provee el concepto de fuente de corriente deprimida que puede llevar a bloqueo en la propagación y terminar la reentrada de forma funcional (43, 44).
Interacciones funcionales del tejido, después de la estimulación prematura (alterando la dispersión en los tres componentes celulares modelados), con obstáculos funcionales como la lesión isquémica (depresión de excitabilidad y por tanto menos corriente disponible en el frente de onda) bloquean la conducción del frente y generan, en la interface de los tejidos, despolarización de manera transversal, que crea la singularidad de fase (45). La zona de borde por donde se propaga el frente reentrante genera un frente convexo que cambia en su trasegar por la zona isquémica; amplios frentes que llegan a la zona distal de la lesión cambian, dependiendo de las condiciones isquémicas, a estrechos y lentos, generando el amplio retraso en la conducción por la zona isquémica y alterando la dispersión de repolarización del tejido. En la reentrada lobular, el concepto clave es la curvatura del frente de onda. Esta curvatura en el frente forma una región de desbalance de alta impedancia (desbalance de fuente-sumidero), donde la corriente provista por el frente reentrante (fuente) es insuficiente para cargar la capacidad del tejido circundante y de este modo excitar largos volúmenes de tejido próximo (sumidero) (46, 47).
Rol de dispersión de repolarización (temporal y espacial)
La duración del periodo refractario es determinada por componentes dependientes de voltaje y tiempo. La duración de la fase de meseta del potencial de acción determina la componente dependiente de voltaje del periodo refractario, mientras que el potencial de reposo determina la componente dependiente de tiempo. El periodo refractario se hace más largo excediendo la duración del potencial reflejando el continuo aumento de hiperkalemia actuando sobre el potencial de reposo y, a través de este mecanismo, alterando la excitabilidad de la membrana (25, 48).
La dispersión no uniforme de refractariedad es referida como los cambios en velocidad de recuperación de excitabilidad punto a punto dentro de una región determinada. La dispersión uniforme hace referencia a cambios monótonos de la refractariedad en aproximadamente la misma cantidad sobre una distancia dada en todas las direcciones y en todos los puntos (49). Cuando se dispersa de manera no uniforme, la refractariedad cambia a diferentes velocidades en diferentes sitios y direcciones de manera no monótona (50, 51). De acuerdo con la hipótesis anterior, el estímulo prematuro aplicado al tejido forma un frente de onda que se bloquea cuando encuentra una región en la cual la dispersión de la refractariedad es altamente no uniforme. El frente de activación no puede desplazarse desde sitios con alta excitabilidad a otros adyacentes de baja excitación y un camino alterno, vía despolarización transversal, sobreviene al bloqueo unidireccional en la interface epicardio-zona lesionada. En caninos se ha mostrado que la secuencia de activación, en especial el protocolo de campo gemelo con CI incremental en la zona refractaria del estímulo básico, distorsiona espacialmente el gradiente de repolarización y genera in-homogeneidades en el tejido que sirven de sustrato para eventos reentrantes (52). En las simulaciones realizadas se nota una marcada alteración de vulnerabilidad si la distribución geométrica de las células M diseñada, compromete la zona de la lesión isquémica. Lo anterior indica que alteraciones dinámicas entre los pulsos aplicados y los frentes reentrantes cambian la secuencia de repolarización de todo el tejido de manera diferencial en diversos puntos del tejido (53).
Rol de heterogeneidad transmural
La heterogeneidad transmural de la pared cardiaca es un parámetro inherente que cambia la disposición celular del modelo diseñado. La alteración del sustrato la genera la existencia diferenciada de células de epicardio, medio miocardio y endocardio con disímiles respuestas eléctricas y distribuidas en distintas zonas, de tamaños y formas variables (54, 55). Gradientes de voltaje transmural aparecen debido a la disparidad de (Ito) en epicardio pero no en endocardio; la expresión génica selectiva potencia esta disparidad transmural en la formación de canales iónicos diferenciados en la pared transmural (56, 57). La diferencia en el transcurso del tiempo de repolarización entre la fase dos y tres en los diferentes tipos de célula forma gradientes opuestos en ambos lados de la región del medio miocardio siendo responsables en gran parte de la inscripción de la onda T del electrocardiograma (58, 59). En las simulaciones, en el límite inferior de intervalo de acople para aplicar el estímulo prematuro, en la interface del tejido epicardio y células M existe un desbalance de excitabilidad muy alto (fotograma 3 en la figura 3). Éste vuelve más lento el discurrir del frente de onda haciendo más difícil, a medida que aumenta el gradiente de hiperkalemia, la propagación por la zona lesionada aumentando el valor del pico de despolarización en el electrograma y variando la duración de la onda S del pulso reentrante (ver electrogramas en la figura 4) (60, 61).
Recientemente se ha encontrado una distribución en forma de islas de células M en pared transmural humana, utilizando métodos ópticos de alto detalle de descripción. Estas distribuciones están de acuerdo con las simulaciones que se presentan en este artículo, lo cual permite su respectiva convalidación experimental en corazones humanos (62).
Rol de hiperkalemia
Los efectos electrofisiológicos del incremento en la concentración de potasio extracelular son el resultado de una alteración en el periodo refractario y por consiguiente sus efectos sobre viabilidad de sodio, potencial máximo de subida y cambios en la conductancia de potasio que afectan la duración de la fase de meseta del potencial y el desarrollo de despolarización espontánea. Las alteraciones en la conductancia de potasio tienen inferencia de manera diferencial en las diferentes corrientes que manejan la fase de meseta del potencial y por ende su duración. En la corriente Ito e IKs puede notarse una marcada diferenciación de estos efectos, que manejan la forma de espiga y domo del potencial y su duración respectivamente (1, 63).
Los cambios en potasio extracelular en la pared ventricular incrementan excitabilidad y refractariedad. El cambio temporal del periodo refractario efectivo a nivel de las células del tejido se debe casi por completo al aumento del potasio extracelular; como resultado, la corriente de sodio dependiente de voltaje se inactiva y la amplitud del potencial decae al igual que el potencial de reposo que pasa de -85 a -62 mV (64). Estos cambios proceden disminuyendo la velocidad de conducción en todo el tejido y reduciendo la viabilidad de corriente para excitar porciones extensas del frente de onda (65). Las alteraciones de potasio extracelular dentro de la zona isquémica se modelaron con un gradiente de ascenso constante, no obstante existe una marcada variabilidad dentro de la zona lesionada en diversos animales (66). Se observó que los niveles de potasio extracelular superiores a 11 mM potenciaban la reentrada con el modelo diseñado (parte A de la figura 5). Datos experimentales de corazones de cerdo sometidos a isquemia regional fijan este aumento de inducción de reentrada y posterior paso a fibrilación entre 8 y 13,5 mM de potasio extracelular en la zona isquémica (67).
El comportamiento de las reentradas obtenidas, respecto de la concentración de potasio extracelular, permite observar un cambio no lineal en su afección electrofisiológica; es decir cambios superiores a 11 mM producen bloqueo unidireccional en la zona proximal y una conducción cada vez más lenta del frente reentrante por la zona isquémica, alterando la recuperación del tejido (parte inicial de la función de vulnerabilidad, figura 4), pero a partir de 12 mM se presenta bloqueo bidireccional (parte final de dichas funciones) en la parte distal de la lesión para todos los intervalos de acople utilizados. En cobayas la hiperkalemia modifica el potencial de reposo, la amplitud del potencial y el APD de manera diferencial, siendo más acentuados en epicardio respecto del endocardio (68). A mayor incremento de la hiperkalemia, más se acrecentó la disparidad de respuesta de las células del epicardio respecto del endocardio (69). En las simulaciones de pared transmural, el gradiente espacial de aumento de potasio extracelular hace que la porción del epicardio sea la más comprometida por los cambios de la isquemia. El bloqueo unidireccional sucede en la interface de la zona de borde y la lesión en células de epicardio.
Rol de acidosis
En cobayas, los efectos de la acidosis sobre disminución de la velocidad de conducción fueron potenciados a concentraciones extracelulares de potasio de 9 a 13 mM. De datos experimentales se conoce la relación intrínseca entre acidosis y decrecimiento en la excitabilidad celular; esta relación puede ser directa a través de la afección de las corrientes de entrada que soportan la excitabilidad (sodio y calcio a través de los canales tipo L) o puede ser indirecta debido a que la acidosis afecta el potencial de reposo y por medio de éste la excitabilidad celular (70).
Los cambios en el potencial de reposo y el potencial de acción producidos por acidosis en isquemia regional pueden actuar en detrimento de favorecer las arritmias reentrantes. El volverse menos negativo el potencial de reposo observado durante acidosis puede contribuir a la depresión del segmento TQ del electrograma (en forma notoria en la figura 4, en su parte inicial los electrogramas hallados siempre presentan dicha deflexión). En la pared transmural canina, la refractariedad es prolongada (debido a retardo en la recuperación de la corriente de sodio) y la conducción es retardada como consecuencia del descenso de la corriente de excitación (20). Utilizando corazones de cobayas y simulaciones a través del modelo de Luo-Rudy se ha mostrado que alteraciones de la corriente (IK1) modifican el establecimiento y la estabilidad de reentradas en tejido cardiaco (33, 71). La acidosis influencia dicha corriente acortando dramáticamente la duración del potencial en sitios cercanos con la zona de bloqueo unidireccional, conformando un ingrediente dinámico adicional que influencia el mantenimiento del circuito reentrante en la zona isquémica.
Rol de la hipoxia
Uno de los mayores efectores de arritmias en eventos isquémicos es la activación de los canales en la sarcolema de potasio activados por ATP debido a su gran influencia en el acortamiento de la duración del potencial en la zona lesionada (35, 72). Sin embargo, breves periodos de isquemia de cero flujo en modelos de animales con activación de la corriente IKATP, presentan protección antiarrítmica y disminuyen el tamaño del infarto (73).
Se ha propuesto que la activación de la corriente IKATP durante los primeros instantes del evento isquémico acorta el potencial y de ahí que exista mucho menos tiempo viable para entrada de calcio dentro de la célula durante la meseta del potencial. En la zona de borde entre la lesión y el tejido sano, el potasio se difunde de territorio isquémico a normal (74, 75); entonces, hacia el final de la zona, donde el potasio puede aproximarse a valores normales, la refractariedad puede corresponder con el APD, y el acelerado acortamiento del APD durante isquemia será acompañado por un correspondiente acortamiento en la refractariedad, el cual puede ser pro-arrítmico (66, 76). Se hace importante notar que es en la zona de borde donde se generan la mayoría de arritmias isquémicas, tanto en modelos animales (19) como en humanos (77, 78).
Aun no está establecida la razón por la cual la activación de IKATP puede presentar efectos duales, en tejido isquémico, esto es arrítmico y pro-arrítmico. Algunas explicaciones pueden basarse en diferencias de especies animales estudiadas. Se han observado efectos anti-arrítmicos en perros (79) y cobayas (17) y efectos pro-arrítmicos en cerdos (15) y conejos (80). Así mismo, el curso del tiempo de pos-repolarización en la refractariedad varía de forma marcada entre especies (81). En las simulaciones realizadas se encontró que la activación de la corriente IKATP acelera la propagación del frente de onda y hace más marcada la alteración temporal en la refractariedad, debido a la repolarización temprana en la zona isquémica del epicardio y, por tanto, un aumento en el periodo refractario (Henao y colaboradores, artículo enviado para publicación). Este aumento bloquea el frente de onda reentrante en la parte distal del tejido y difumina la propagación del potencial, impidiendo de esta manera el establecimiento de una reentrada funcional en el tejido (82).
Comportamiento de vulnerabilidad
La vulnerabilidad de la pared del miocardio en diferentes minutos de isquemia regional es desconocida y la influencia de diversos parámetros (dispersión transmural de repolarización, componentes de la isquemia) es primordial para comprender la fenomenología de iniciación y desestabilización de frentes reentrantes. Un efecto importante es el que sucede cuando el estímulo prematuro genera un potencial que se propaga de manera exitosa en un tejido que relativamente aun no se ha recuperado. La velocidad de propagación del frente de onda es necesariamente más baja que aquella predecesora, por dos razones. Primero, la velocidad en el medio que se ha recuperado parcialmente es más baja debido a que las células vecinas de manera lateral no son estimuladas por el frente de onda que se mueve, sino que actúan como sumidero de corriente, deprimiendo la velocidad de conducción. Segundo, eventualmente, si el medio es lo suficientemente grande, la porción local de medio estimulada por el frente de onda no es muy grande en la fase vulnerable, entonces la propagación lateral comienza a ser posible (83). En este punto la propagación se convierte en retrógrada; el frente de onda se mueve hacia atrás a través de la vecindad por un camino viable de tejido recuperado hacia el punto de estimulación original (84).
La dispersión no-uniforme de refractariedad, la cual es amplificada por eventos bioquímicos en isquemia, se reconoce como uno de los mayores factores de inducción de arritmias letales en miocardio cardiaco. Esta dispersión forma el sustrato para bloqueo unidireccional de conducción, indispensable para la formación de eventos reentrantes mediados por estimulación prematura (extrasístole ventricular). En estos términos, vemos que la dispersión no uniforme de refractariedad es una condición necesaria (Figura 4) para la predisposición a arritmias en la pared transmural cardiaca. Aunque la cuantificación de este parámetro es un hecho estadístico, en las funciones que se reportan en este trabajo, la reentrada es muchísimo más probable en presencia de distribución geométrica de células M que comprometen la zona de la lesión (aumentando el gradiente de dispersión transmural de repolarización) y se establecen cuando la hiperkalemia y la acidosis logran su mayor valor en el minuto 8,75 de isquemia (este comportamiento se mantiene para todas las distribuciones de células M diseñadas, como se observa en la figuras 4 y 5).
En cobayas se ha cuantificado la influencia de dispersión espacial de repolarización y su influencia en la vulnerabilidad a arritmias en presencia de obstáculos anatómicos (85). En modelos de síndrome de LQT, se ha medido utilizando métodos ópticos la alternación de la onda T y el gradiente transmural de repolarización y su interdependencia con vulnerabilidad a reentrada (86). En este trabajo la dispersión de repolarización conforma la base para el establecimiento de elevación del segmento ST y depresión TQ en los electrogramas obtenidos y concuerda con resultados experimentales (87).
Conclusión
La vulnerabilidad a reentrada en la pared transmural sujeta a isquemia regional, sin variar el punto de aplicación del estímulo y la amplitud de la corriente aplicada, depende en este caso de dos tipos de gradientes presentes de manera funcional en la configuración de tejido modelada:
- Gradientes permanentes de parámetros (hiperkalemia, acidosis, heterogeneidad celular).
- Gradientes efímeros de estados (velocidad de conducción, curvatura, dispersión transmural de repolarización).
El delicado balance de gradientes morfológicos transmurales y funcionales creados por la isquemia en el tejido, sirve como formante mediador de un comportamiento dinámico y parada autónoma de eventos reentrantes.
La heterogeneidad bioquímica y morfológica en el tejido virtual estudiado da como resultado una arritmia por reentrada; su influencia en la vulnerabilidad del tejido aumenta a medida que la severidad de la hiperkalemia crece. Los electrogramas obtenidos presentan depresión TQ y elevación ST con una morfología de taquicardia ventricular polimórfica.
Agradecimientos
Este trabajo fue parcialmente financiado por el Plan Nacional de Investigación Científica, Desarrollo e Innovación Tecnológica del Ministerio de Educación y Ciencia (proyectos TIN2004-03602 y TEC 2005-04199/TCM), de España.
Bibliografía
1. Antzelevitch C, Sicouri S, Litovsky SH, et al. Heterogeneity within the ventricular wall. Electrophysiology and pharmacology of epicardial, endocardial, and M cells. Circ Res 1991; 69 (6): 1427-1449. [ Links ]
2. Litovsky SH, Antzelevitch C. Transient outward current prominent in canine ventricular epicardium but not endocardium. Circ Res 1988; 62 (1): 116-126. [ Links ]
3. Sicouri S, Antzelevitch C. A subpopulation of cells with unique electrophysiological properties in the deep subepicardium of the canine ventricle. The M cell. Circ Res 1991; 68 (6): 1729-1741. [ Links ]
4. Antzelevitch C. Modulation of transmural repolarization. Ann N Y Acad Sci 2005; 1047: 314-323. [ Links ]
5. Liu DW, Gintant GA, Antzelevitch C. Ionic bases for electrophysiological distinctions among epicardial, midmyocardial, and endocardial myocytes from the free wall of the canine left ventricle. Circ Res 1993; 72 (3): 671-687. [ Links ]
6. Yan GX, Lankipalli RS, Burke JF, Musco S, Kowey PR. Ventricular repolarization components on the electrocardiogram: cellular basis and clinical significance. J Am Coll Cardiol 2003; 42 (3): 401-409. [ Links ]
7. Gima K, Rudy Y. Ionic current basis of electrocardiographic waveforms: a model study. Circ Res 2002; 90 (8): 889-896. [ Links ]
8. Antzelevitch C. Heterogeneity and cardiac arrhythmias: an overview. Heart Rhythm 2007; 4 (7): 964-972. [ Links ]
9. Antzelevitch C. Role of spatial dispersion of repolarization in inherited and acquired sudden cardiac death syndromes. Am J Physiol Heart Circ Physiol 2007; 293 (4): H2024-H2038. [ Links ]
10. Atar S, Barbagelata A, Birnbaum Y. Electrocardiographic diagnosis of ST-elevation myocardial infarction. Cardiol Clin 2006; 24 (3): 343-65, VII. [ Links ]
11. Antzelevitch C, Fish J. Electrical heterogeneity within the ventricular wall. Basic Res Cardiol 2001; 96 (6): 517-527. [ Links ]
12. Antzelevitch C. Cardiac repolarization. The long and short of it. Europace. 2005; 7 (Suppl 2): 3-9. [ Links ]
13. Coronel R, Fiolet JW, Wilms-Schopman FJ, et al. Distribution of extracellular potassium and its relation to electrophysiologic changes during acute myocardial ischemia in the isolated perfused porcine heart. Circulation 1988; 77 (5): 1125-1138. [ Links ]
14. Janse MJ, Cinca J, Morena H et al. The "border zone" in myocardial ischemia. An electrophysiological, metabolic, and histochemical correlation in the pig heart. Circ Res 1979; 44 (4): 576-588. [ Links ]
15. Morena H, Janse MJ, Fiolet JW et al. Comparison of the effects of regional ischemia, hypoxia, hyperkalemia, and acidosis on intracellular and extracellular potentials and metabolism in the isolated porcine heart. Circ Res 1980; 46 (5): 634-646. [ Links ]
16. Cascio WE, Johnson TA, Gettes LS. Electrophysiologic changes in ischemic ventricular myocardium: I. Influence of ionic, metabolic, and energetic changes. J Cardiovasc Electrophysiol 1995; 6 (11): 1039-1062. [ Links ]
17. Wilde AA, Kleber AG. The combined effects of hypoxia, high K+, and acidosis on the intracellular sodium activity and resting potential in guinea pig papillary muscle. Circ Res 1986; 58 (2): 249-256. [ Links ]
18. Wilde AA, Aksnes G. Myocardial potassium loss and cell depolarisation in ischaemia and hypoxia. Cardiovasc Res 1995; 29 (1): 1-15. [ Links ]
19. Janse MJ, Wit AL. Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischemia and infarction. Physiol Rev 1989; 69 (4): 1049-1169.
20. Carmeliet E. Cardiac ionic currents and acute ischemia: from channels to arrhythmias. Physiol Rev 1999; 79 (3): 917-1017. [ Links ] [ Links ]
21. Ferrero JM, Jr., Saiz J, Ferrero JM, Thakor NV. Simulation of action potentials from metabolically impaired cardiac myocytes. Role of ATP-sensitive K+ current. Circ Res 1996; 79 (2): 208-221. [ Links ]
22. Dumaine R, Towbin JA, Brugada P, et al. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res 1999; 85 (9): 803-809. [ Links ]
23. Viswanathan PC, Shaw RM, Rudy Y. Effects of IKr and IKs heterogeneity on action potential duration and its rate dependence: a simulation study. Circulation 1999; 99 (18): 2466-2474. [ Links ]
24. Kleber AG, Janse MJ, Wilms-Schopmann FJ, Wilde AA, Coronel R. Changes in conduction velocity during acute ischemia in ventricular myocardium of the isolated porcine heart. Circulation 1986; 73 (1): 189-198. [ Links ]
25. Cascio WE. Myocardial ischemia: what factors determine arrhythmogenesis? J Cardiovasc Electrophysiol 2001; 12 (6): 726-729. [ Links ]
26. Faber GM, Rudy Y. Action potential and contractility changes in [Na(+)](i) overloaded cardiac myocytes: a simulation study. Biophys J 2000; 78 (5): 2392-2404. [ Links ]
27. Luo CH, Rudy Y. A dynamic model of the cardiac ventricular action potential. I. Simulations of ionic currents and concentration changes. Circ Res 1994; 74 (6): 1071-1096. [ Links ]
28. Rudy Y, Silva JR. Computational biology in the study of cardiac ion channels and cell electrophysiology. Q Rev Biophys 2006; 39 (1): 57-116. [ Links ]
29. Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol 1952; 117 (4): 500-544. [ Links ]
30. Weidmann S. Electrical constants of trabecular muscle from mammalian heart. J Physiol 1970; 210 (4): 1041-1054. [ Links ]
31. Jongsma HJ, Wilders R. Gap junctions in cardiovascular disease. Circ Res 2000; 86 (12): 1193-1197. [ Links ]
32. Kleber AG, Rudy Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol Rev 2004; 84 (2): 431-488. [ Links ]
33. Beaumont J, Davidenko N, Davidenko JM, Jalife J. Spiral waves in two-dimensional models of ventricular muscle: formation of a stationary core. Biophys J 1998; 75 (1): 1-14. [ Links ]
34. Coronel R. Heterogeneity in extracellular potassium concentration during early myocardial ischaemia and reperfusion: implications for arrhythmogenesis. Cardiovasc Res 1994; 28 (6): 770-777. [ Links ]
35. Ferrero JM, Jr., Trenor B, Rodríguez B, Sáiz J. Electrical activity and reentry during acute regional myocardial ischemia: insights from simulations. Int J Bifurcation and Chaos 2003; 13 (12): 3703-3715. [ Links ]
36. Liu DW, Antzelevitch C. Characteristics of the delayed rectifier current (IKr and IKs) in canine ventricular epicardial, midmyocardial, and endocardial myocytes. A weaker IKs contributes to the longer action potential of the M cell. Circ Res 1995; 76 (3): 351-365. [ Links ]
37. Clayton RH, Holden AV. Propagation of normal beats and re-entry in a computational model of ventricular cardiac tissue with regional differences in action potential shape and duration. Prog Biophys Mol Biol 2004; 85 (2-3): 473-499. [ Links ]
38. Akar FG, Yan GX, Antzelevitch C, Rosenbaum DS. Unique topographical distribution of M cells underlies reentrant mechanism of torsade de pointes in the long-QT syndrome. Circulation 2002; 105 (10): 1247-1253. [ Links ]
39. Poelzing S, Rosenbaum DS. Nature, significance, and mechanisms of electrical heterogeneities in ventricle. Anat Rec A Discov Mol Cell Evol Biol 2004; 280 (2): 1010-1017. [ Links ]
40. Henao O, Ferrero JM, Ramírez E, Sáiz J. Arritmias cardiacas generadas por heterogeneidad electrofisiológica: estudio mediante simulación. Rev Colomb Cardiol 2007; 14 (4): 185-197. [ Links ]
41. Geselowitz DB. On the theory of the electrocardiogram. Proceedings of the IEEE 1989; 77 (6): 857-876. [ Links ]
42. Saiz J, Ferrero JM, Jr., Monserrat M, Ferrero JM, Thakor NV. Influence of electrical coupling on early afterdepolarizations in ventricular myocytes. IEEE Trans Biomed Eng 1999; 46 (2): 138-147. [ Links ]
43. Winfree AT. On measuring curvature and electrical diffusion coefficients in anisotropic myocardium: comments on "effects of bipolar point and line simulation in anisotropic rabbit epicardium: assessment of the critical radius of curvature for longitudinal block". IEEE Trans Biomed Eng 1996; 43 (12): 1200-1203. [ Links ]
44. Winfree AT. Electrical singular filaments in the heart wall. In: The Geometry of Biological Time, 2nd. ed. J. Marsden, L. Sirovich, and S. Wiggins, Eds. Springer Verlag; 2001. p. 455-526. [ Links ]
45. Sidorov VY, Aliev RR, Woods MC, et al. Spatiotemporal dynamics of damped propagation in excitable cardiac tissue. Phys Rev Lett 2003; 91 (20): 208104. [ Links ]
46. Winfree AT. Heart muscle as a reaction-diffusion medium: The roles of electric potential diffusion, activation front curvature, and anisotropy. Int J Bifurcation and Chaos 1997; 7 (3): 487-526. [ Links ]
47. Fast VG, Kleber AG. Role of wavefront curvature in propagation of cardiac impulse. Cardiovasc Res 1997; 33 (2): 258-271. [ Links ]
48. Gettes LS, Cascio WE. Effect of acute ischemia on cardiac electrophysiology. In: The Heart and Cardiovascular System. H. A. Fozzard, E. Haber, R. Jennings, A. Katz, and H. Morgan, Eds. New York: Raven Press ltd.; 1992. p. 2021-2053. [ Links ]
49. Franz MR, Zabel M. Electrophysiological basis of QT dispersion measurements. Prog Cardiovasc Dis 2000; 42 (5): 311-324. [ Links ]
50. Burton FL, Cobbe SM. Dispersion of ventricular repolarization and refractory period. Cardiovasc Res 2001; 50 (1): 10-23. [ Links ]
51. Restivo M, Gough WB, El-Sherif N. Ventricular arrhythmias in the subacute myocardial infarction period. High-resolution activation and refractory patterns of reentrant rhythms. Circ Res 1990; 66 (5): 1310-1327. [ Links ]
52. Osaka T, Kodama I, Tsuboi N, Toyama J, Yamada K. Effects of activation sequence and anisotropic cellular geometry on the repolarization phase of action potential of dog ventricular muscles. Circulation 1987; 76 (1): 226-236. [ Links ]
53. Restivo M, Caref EB, Kozhevnikov DO, El-Sherif N. Spatial dispersion of repolarization is a key factor in the arrhythmogenicity of long QT syndrome. J Cardiovasc Electrophysiol 2004; 15 (3): 323-331. [ Links ]
54. Akar FG, Yan GX, Antzelevitch C, Rosenbaum DS. Unique topographical distribution of M cells underlies reentrant mechanism of torsade de pointes in the long-QT syndrome. Circulation 2002; 105 (10): 1247-1253. [ Links ]
55. Yan GX, Shimizu W, Antzelevitch C. Characteristics and distribution of M cells in arterially perfused canine left ventricular wedge preparations. Circulation 1998; 98 (18): 1921-1927. [ Links ]
56. Gaborit N, Le Bouter S, Szuts V, et al. Regional and tissue specific transcript signatures of ion channel genes in the non-diseased human heart. J Physiol 2007; 582 (2): 675-693. [ Links ]
57. Soltysinska E, Olesen S, Christ T et al. Transmural expression of ion channels and transporters in human nondiseased and end-stage failing hearts. Pflugers Archiv Eur J Physiol 2009; 459 (1): 11-23. [ Links ]
58. Antzelevitch C, Shimizu W, Yan GX et al. The M cell: Its contribution to the ECG and to normal and abnormal electrical function of the heart. J Cardiovasc Electrophysiol 1999; 10 (8): 1124-1152. [ Links ]
59. Antzelevitch C. Cellular basis for the repolarization waves of the ECG. Ann N Y Acad Sci 2006; 1080: 268-281. [ Links ]
60. Janse MJ. ST segment mapping and infarct size. Cardiovasc Res 2000; 45 (1): 190-193. [ Links ]
61. Janse MJ. ST-segment elevation or TQ-segment depression? Heart Rhythm 2007; 4 (2): 207. [ Links ]
62. Glukhov A, Fedorov V, Lou Q et al. Transmural dispersion of repolarization in failing and nonfailing human ventricle. Circ Res 2010; 106 (5): 981-991. [ Links ]
63. Burashnikov A, Antzelevitch C. Prominent I(Ks) in epicardium and endocardium contributes to development of transmural dispersion of repolarization but protects against development of early afterdepolarizations. J Cardiovasc Electrophysiol 2002; 13 (2): 172-177. [ Links ]
64. Kleber AG. Resting membrane potential, extracellular potassium activity, and intracellular sodium activity during acute global ischemia in isolated perfused guinea pig hearts. Circ Res 1983; 52 (4): 442-450. [ Links ]
65. Kleber AG, Janse MJ, Wilms-Schopmann FJ, Wilde AA, Coronel R. Changes in conduction velocity during acute ischemia in ventricular myocardium of the isolated porcine heart. Circulation 1986; 73 (1): 189-198. [ Links ]
66. Tice BM, Rodríguez B, Eason J, Trayanova N. Mechanistic investigation into the arrhythmogenic role of transmural heterogeneities in regional ischaemia phase 1A. Europace 2007; 9 (Suppl 6): VI46-VI58. [ Links ]
67. Coronel R, Wilms-Schopman FJ, Dekker LR, Janse MJ. Heterogeneities in [K+] o and TQ potential and the inducibility of ventricular fibrillation during acute regional ischemia in the isolated perfused porcine heart. Circulation 1995; 92 (1): 120-129. [ Links ]
68. Wan X, Bryant SM, Hart G. The effects of [K+]o on regional differences in electrical characteristics of ventricular myocytes in guinea-pig. Exp. Physiol 2000; 85 (6): 769-774. [ Links ]
69. Gronich N, Kumar A, Zhang Y, Efimov I, Soldatov N. Molecular remodeling of ion channels, exchangers and pumps in atrial and ventricular myocytes in ischemic cardiomyopathy. Channels 2010; 4 (2): 1-7. [ Links ]
70. Kagiyama Y, Hill JL, Gettes LS. Interaction of acidosis and increased extracellular potassium on action potential characteristics and conduction in guinea pig ventricular muscle. Circ Res 1982; 51 (5): 614-623. [ Links ]
71. Jalife J, Berenfeld O. Molecular mechanisms and global dynamics of fibrillation: an integrative approach to the underlying basis of vortex-like reentry. J Theor Biol 2004; 230 (4): 475-487. [ Links ]
72. Ferrero J, Jr., Torres V, Montilla F, Colomar E. Simulation of reentry during acute myocardial ischemia: role of ATP-sensitive potassium current and acidosis. Computers in Cardiology 2000; 27: 239-242. [ Links ]
73. Remme CA, Wilde AA. KATP channel openers, myocardial ischemia, and arrhythmias -should the electrophysiologist worry? Cardiovasc. Drugs Ther 2000; 14 (1): 17-22. [ Links ]
74. Taggart P, Yellon DM. Preconditioning and arrhythmias. Circulation 2002; 106 (24): 2999-3001. [ Links ]
75. Yan GX, Yamada KA, Kleber AG, McHowat J, Corr PB. Dissociation between cellular K+ loss, reduction in repolarization time, and tissue ATP levels during myocardial hypoxia and ischemia. Circ Res 1993; 72 (3): 560-570 [ Links ]
76. Trenor B, Romero L, Ferrero JM, Jr., et al. Vulnerability to reentry in a regionally ischemic tissue: a simulation study. Ann Biomed Eng 2007; 35 (10): 1756-1770. [ Links ]
77. Taggart P, Sutton PM, Opthof T et al. Transmural repolarisation in the left ventricle in humans during normoxia and ischaemia. Cardiovasc Res 2001; 50 (3): 454-462.< [ Links ]
78. Sutton PMI, Taggart P, Opthof T et al. Repolarisation and refractoriness during early ischaemia in humans. Heart 2000; 84 (4): 365-369. [ Links ]
79. Donaldson RM, Nashat FS, Noble D, Taggart P. Differential effects of ischaemia and hyperkalaemia on myocardial repolarization and conduction times in the dog. J Physiol 1984; 353: 393-403. [ Links ]
80. Weiss JN, Venkatesh N, Lamp ST. ATP-sensitive K+ channels and cellular K+loss in hypoxic and ischaemic mammalian ventricle. J Physiol 1992; 447: 649-673. [ Links ]
81. Janse MJ, van Capelle FJ, Morsink H, et al. Flow of "injury" current and patterns of excitation during early ventricular arrhythmias in acute regional myocardial ischemia in isolated porcine and canine hearts. Evidence for two different arrhythmogenic mechanisms. Circ Res 1980; 47 (2): 151-165. [ Links ]
82. Glukhov A, Flagg T, Fedorov V, Efimov I, Nichols C. Differential KATP channel pharmacology in intact mouse heart. J Molec Cell Cardiol 2010; 48 (1): 152-160.
83. Keener J. A mathematical model for the vulnerable phase in myocardium. Mathematical Biosciences 1988; 90 (1): 3-18. [ Links ] [ Links ]
84. Winfree AT. Electrical instability in cardiac muscle: phase singularities and rotors. J Theor Biol 1989; 138 (3): 353-405 [ Links ]
85. Laurita KR, Girouard SD, Akar FG, Rosenbaum DS. Modulated dispersion explains changes in arrhythmia vulnerability during premature stimulation of the heart. Circulation 1998; 98 (24): 2774-2780. [ Links ]
86. Wan X, Laurita KR, Pruvot EJ, Rosenbaum DS. Molecular correlates of repolarization alternans in cardiac myocytes. J Mol Cell Cardiol 2005; 39 (3): 419-428. [ Links ]
87. Di Diego JM, Antzelevitch C. Cellular basis for ST-segment changes observed during ischemia. J Electrocardiol 2003; 36 (Suppl): 1-5. [ Links ]

