










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Cardiología
Print version ISSN 0120-5633
Rev. Colom. Cardiol. vol.18 no.2 Bogota Mar./Apr. 2011
Mecanismos de cardiotoxicidad: antineoplásicos, anti-inflamatorios no esteroideos, antipsicóticos, cocaetileno y simpaticomiméticos
Mechanisms of cardiotoxicity: antineoplastics, nonsteroidal anti-inflammatory drugs, antipsychotics, cocaethylene and sympathomimetics
(1) Fundación Santa Fe de Bogotá, Bogotá, Colombia.
Correspondencia: Dr. Lukas Salazar Sierra. Cra. 69 D No. 24 C-50 Bloque 2 Apto. 803. Bogotá, DC. Colombia. Tel.: (57-1) 8 01 72 71 - 301 750 70 09- 300 551 71 00. Correo electrónico: lukassal@hotmail.com
Recibido: 21/05/2010. Aceptado: 13/12/2010.
La interacción constante del organismo humano con diferentes sustancias, que incluso en muchas ocasiones se consideran inofensivas, tiene un alto impacto sobre todos los sistemas, siendo el cardiovascular uno de los más afectados. Por lo tanto, es vital reconocer los mecanismos por los cuales estas sustancias ejercen su efecto tóxico sobre este sistema, bien sea afectando la estabilidad de membrana y la función contráctil o generando disfunción de organelos intracelulares y estrés oxidativo. Numerosos estudios han descubierto efectos lesivos de sustancias, como la clozapina y las catecolaminas, que han tenido amplio uso durante largos años. En la actualidad aún se realizan investigaciones que buscan esclarecer los mecanismos cardiotóxicos de medicamentos de formulación común, entre ellos antineoplásicos y anti-inflamatorios no esteroideos (AINE), así como de sustancias de uso habitual que causan adicción, tales como alcohol, cocaína y cocaetileno, su metabolito activo.
Palabras clave: cardiotoxicidad, arritmogénesis, daño miocárdico, antineoplásicos, anti-inflamatorios no esteroideos, cocaetileno, clozapina, catecolaminas.
The constant interaction of the human body with different substances that are even in many cases considered harmless has a high impact on all systems, being the cardiovascular system one of the most affected. Therefore, it is vital to recognize the mechanisms by which these substances exert their toxic effect on this system, either affecting the membrane stability and the contractile function, or generating intracellular organelles dysfunction and oxidative stress. Numerous studies have found that drugs which have been widely used for many years such as clozapine and catecholamines, have harmful effects. Research is still being done seeking to clarify the cardiotoxic mechanisms of drugs commonly formulated, including anticancer and non steroidal anti-inflammatory drugs (NSAIDs), as well as commonly used substances that cause addiction, such as alcohol, cocaine and cocaethylene, its active metabolite.
Key words: cardiotoxicity, arrhythmogenesis, myocardial injury, antineoplastics, nonsteroidal anti-inflammatory drugs, cocaethylene, clozapine, catecholamines.
Introducción
Las intoxicaciones conforman uno de los principales motivos de consulta en los servicios de urgencias y a su vez una de las causas más comunes de paro cardiorrespiratorio según la American Heart Association (AHA) (1) y pueden producirse accidentalmente, en acciones delictivas o incluso voluntarias. Es de suma importancia en el ámbito clínico conocer los mecanismos de daño asociados con las alteraciones cardiacas consecuencia de la exposición a diferentes sustancias y cómo éstas interactúan con los sistemas de contracción y de conducción cardiacas.
Con esta revisión se busca desarrollar procesos de pensamiento y acción que permitan la adecuada atención del paciente intoxicado.
Materiales y métodos
Se realizó una búsqueda sistemática de artículos mediante Pubmed, teniendo en cuenta palabras clave que aportaran información precisa y suficiente. Adicionalmente, en la base bibliográfica se incluyeron libros de toxicología clínica mediante búsqueda física y referenciada, y algunos artículos de importancia histórica para los temas a tratar.
Para establecer los criterios de inclusión, se utilizó el mecanismo de «Límites» del buscador Pubmed, en los que se tuvieron en cuenta:
- Fecha de publicación: entre 2000/01/01 y 2009/09/30.
- Lenguaje: español o inglés.
- Tipo de artículo: ensayo clínico, ensayo controlado aleatorizado, meta-análisis o revisión sistemática de la literatura.
- Vínculos a texto completo.
Al momento de realizar la exclusión de artículos, se valoró la idoneidad de las revistas donde se publicaron, y se dio prioridad a las revistas de farmacología, toxicología y cardiología, excluyendo aquellas que no tuvieran relación con estos tópicos.
Efectos de los tóxicos sobre el corazón
Varias sustancias pueden actuar como tóxicos para el corazón mediante diferentes mecanismos que afectan su funcionamiento debido a sus efectos sobre la frecuencia de contracción, ritmo, aporte sanguíneo al miocardio, entre otros. Esto favorece las respuestas de compensación a estímulos nocivos que posteriormente comprometerán la adecuada funcionalidad del sistema cardiovascular.
Entre las alteraciones de la frecuencia y el ritmo cardiaco se encuentran las bradiarritmias y taquiarritmias, que pueden llegar a ser sintomáticas y poner en riesgo la vida del paciente. Se clasifican en supraventriculaes y ventriculares con base en el sitio de su origen. Estas alteraciones pueden diagnosticarse mediante el electrocardiograma, en el que se pueden valorar el ritmo cardíaco, la frecuencia, la duración del complejo QRS y el intervalo QT. Las arritmias supraventriculares generalmente tienen QRS menor de 120 ms y se asocian con mecanismos de reentrada o bypass anatómico como la fibrilación o el aleteo (flutter) auricular. Las arritmias ventriculares se presentan con un QRS mayor de 120 ms y/o QT prolongado; se asocian a procesos isquémicos, cicatriciales e hipertróficos y se pueden manifestar como extrasístoles frecuentes, fibrilación o taquicardia ventricular.
El compromiso a nivel del aporte sanguíneo al miocardio tiene como consecuencia la alteración entre la demanda y la entrega efectiva de oxígeno al sistema, lo que puede causar infarto agudo del miocardio, angina de pecho, angina de Prinzmetal (vasoconstricción coronaria), o una lesión crónica isquémica que posteriormente puede conducir al desarrollo de falla cardiaca.
Al igual que los procesos isquémicos, existen otros estímulos que afectan la funcionalidad del miocardio y que pueden producir finalmente falla cardiaca; estos son: hipertensión arterial sostenida, aumento de la carga mecánica y estímulos hormonales (angiotensina II, a1–adrenérgicos, factores de crecimiento, citoquinas, prostaglandinas, entre otras) los cuales favorecen la remodelación cardiaca hacia la hipertrofia como respuesta compensadora del miocardio mediante el aumento del tamaño celular y de la síntesis proteica (especialmente por la activación de genes precursores de cadenas pesadas de miosina y actina fetal alfa), con la consecuente reducción de la compliance, que a largo plazo lleva a hipoperfusión tisular local, con respuestas compensadoras neurohormonales dadas principalmente por la liberación de péptidos natriuréticos (ANP y BNP), que inducen disfunción celular y apoptosis, lo que precipitará la aparición de miocardiopatía dilatada y falla cardiaca por disminución de la contractilidad y reducción del gasto cardiaco (2, 3).
Mecanismos de cardiotoxicidad
Las sustancias cardiotóxicas ejercen su efecto en diferentes aspectos del funcionamiento cardiaco ya sea de manera intrínseca o extrínseca, alterando la funcionalidad adecuada del sistema. Estos mecanismos de acción tienen como ejes principales el ritmo cardiaco (arritmogénesis), la alteración del flujo sanguíneo coronario, el estrés oxidativo y la disfunción de los organelos celulares que finalmente derivan en procesos de muerte celular (apoptosis y necrosis) o en remodelación miocárdica, compensadora en un comienzo, pero con consecuencias desfavorables a largo plazo.
Mecanismos de arritmogénesis
El ritmo normal del corazón es dirigido por el nódulo sinusal conformado por células que tienen capacidad de estímulo más rápida que el resto del sistema de conducción cardiaca, con frecuencias promedio de 60 a 100 latidos por minuto. El funcionamiento normal del nódulo sinusal y del sistema de conducción cardiaca depende de la adecuada generación y morfología del potencial de acción miocárdico, que comprende cuatro fases: la despolarización rápida dependiente de los canales de sodio (Na+) (fase 0, 1) la meseta dependiente del calcio (Ca+2) (fase 2), la repolarización dependiente de potasio (K+) (fase 3) y el potencial de reposo dependiente de bomba Na+ K+-ATPasa, (fase 4) (Figura 1). Si alguna de éstas se altera, cualquiera de las otras células del sistema de conducción eléctrico cardiaco con capacidad de automatismo, podrá tomar el comando de los estímulos y posesionarse como nuevo marcapasos desplazando al nódulo sinusal de esta tarea y alterando así el ritmo cardiaco (4).
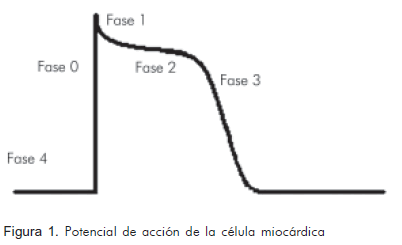
Pueden generarse estímulos espontáneos no inhibidos adecuadamente sobre el potencial de acción, que alcanzan fácilmente el umbral y producen impulsos efectivos sobre las fases 2 a la 4, efecto conocido como actividad gatillada. Estos estímulos se conocen con el nombre de post-despolarizaciones, las cuales pueden ser tempranas o tardías, dependiendo de la fase en la que ocurran o del mecanismo de producción. Las post-despolarizaciones tempranas ocurren durante las fases 2 y 3 y son dadas principalmente por la prolongación del potencial de acción debido al bloqueo de canales de salida de K+ que favorece un aumento en la concentración citoplasmática de este ión, permitiendo así el retraso en el regreso al potencial eléctrico de reposo, lo que facilita la generación de nuevas descargas en este periodo. Las post-despolarizaciones tardías producidas por una sobrecarga de calcio citoplasmático, pueden deberse a la alteración del balance entre las corrientes de salida y entrada de calcio o a sustancias que faciliten la apertura de receptores de rianodina en el retículo sarcoplasmático favoreciendo la salida de calcio hacia el sarcoplasma (5). Las despolarizaciones tardías ocurren durante la fase 4, pero antes de que se genere el impulso normal siguiente (2, 6, 7).
Otro mecanismo de arritmogénesis es producido por conducción anormal del impulso generado de manera correcta, debido a circuitos de reentrada y depresión de la membrana celular. El mecanismo de reentrada está generado por un área refractaria (isquemia, cicatriz, depresión de membrana, etc.) que bloquea la conducción de un impulso, el cual continúa por una vía alterna que posteriormente regresa al área previamente refractaria y si ésta ahora es excitable, se generará un circuito de reentrada. La depresión de membrana describe el efecto que posee sobre el potencial de acción la interacción de los diferentes tóxicos con los canales iónicos que lo causan, como se describe a continuación (6).
Bloqueo de los canales de sodio
Al disminuirse la cantidad de canales de sodio libres requeridos para permitir el inicio del potencial de acción, se hace necesario un estímulo mucho mayor para poder desencadenarlo. Esto es potencialmente favorable para el mecanismo de acción de algunos antiarrítmicos, ya que disminuye el automatismo e inhibe la actividad gatillada (post-despolarizaciones); por otra parte, y como mecanismo adverso de algunas sustancias, favorece el enlentecimiento de la pendiente de despolarización (reflejado en el electrocardiograma como un ensanchamiento del QRS) y consecuentemente de la velocidad de conducción ventricular, lo cual bloquea de forma segmentaria la conducción normal y da paso a la creación de circuitos de reentrada que a ese nivel pueden desembocar en arritmias como taquicardia o fibrilación ventricular (2, 7, 8).
Bloqueo de canales de potasio
Por la función de estos canales en el potencial de acción, su bloqueo conllevará la prolongación de la fase de repolarización, siendo benéfico para la acción de los antiarrítmicos que actúan sobre esta fase al aumentar la refractariedad; pero al actuar como efecto adverso se manifiesta por la prolongación del QT en el electrocardiograma, lo que predispone a fenómenos de reentrada, a la aparición de post-despolarizaciones tempranas y al incremento de la probabilidad de taquicardias ventriculares polimórficas incluida la torsades de pointes (torsión de puntas) con QT mayor a 500 ms (2, 7).
Bloqueo de los canales de calcio
Si bien este mecanismo es favorable en muchos escenarios clínicos, con el uso de medicamentos antagonistas de los canales de calcio es importante conocer los efectos cardiotóxicos que pueden llegar a tener en eventos tales como la sobredosificación. Las principales acciones tóxicas cardiacas, dadas fundamentalmente por la acentuación de su acción farmacológica, las poseen las sustancias no dihidropiridínicas, que disminuyen la velocidad de conducción del impulso con efecto inotrópico y cronotrópico negativos, y, en consecuencia, alteran el gasto cardiaco. Éstas pueden inhibir la despolarización espontánea del nodo sinusal y causar disminución de la conducción a través del nodo aurículo-ventricular (nodo AV) produciendo ritmos de bradicardia, bloqueos AV y ritmos de la unión o idioventriculares. Por otra parte, cuando las sustancias dihidropiridínicas intensifican su acción farmacológica, producen hipotensión arterial por vasodilatación y taquicardia refleja (2, 7, 8).
Bloqueo de la bomba Na+K+ATPasa
Produce aumento en la concentración de sodio citoplasmático que a su vez activa la bomba Na+/Ca+2; esto incrementará la concentración de calcio citoplasmático causando efecto inotrópico positivo, característica farmacológica de algunos medicamentos como los digitálicos. Los cambios electrocardiográficos normales de la impregnación digitálica se caracterizan por ondas T aplanadas, depresión del segmento ST y acortamiento del intervalo QT. Los efectos tóxicos de dichos medicamentos se basan en el aumento del automatismo de las células cardiacas, el enlentecimiento de la conducción a través del nodo AV y la producción de post-despolarizaciones tardías que dan lugar a numerosas arritmias, usualmente asociadas a hiperkalemia, con los cambios electrocardiográficos consecuentes (ondas T picudas y ensanchamiento del complejo QRS) (2, 7, 8).
Los efectos cardiotóxicos se producen no sólo por la acción directa de ciertas sustancias sobre los receptores propios del potencial de acción; éstas pueden actuar también en otros sistemas que secundariamente afectan al cardiovascular, incluyendo aquellas que alteran la función del sistema nervioso autónomo. En el sistema simpático el estímulo sobre los receptores a1 cardiacos favorece la fosforilación de los canales de Ca+2 tipo L durante la despolarización, a la vez que se fosforila el fosfolamban permitiendo la recaptación de Ca+2 por el retículo sarcoplasmático, y se estimula así la contracción y la relajación con un efecto cronotrópico positivo. El bloqueo de estos receptores tendrá como consecuencia un efecto opuesto disminuyendo el automatismo, la contractilidad y la velocidad de conducción del impulso, con prolongación de la conducción a través del nodo AV. La estimulación de receptores nicotínicos del sistema parasimpático, disminuye la excitabilidad del nódulo sinusal y la conducción a través del nodo AV favoreciendo la aparición de bradicardia y de bloqueos aurículo-ventriculares (bloqueos AV) (6).
Es importante anotar que mediante los mecanismos en mención tienen su acción farmacológica algunos medicamentos benéficos para el paciente, así como se observa en la clasificación propuesta por Vaughan–Williams para los antiarrítmicos, donde la clase I corresponde a bloqueadores de canales de sodio, la clase II a beta-bloqueadores, la clase III a bloqueadores de canales de potasio y la clase IV a calcioantagonistas (9). Se debe pues diferenciar entre los efectos farmacológicos deseados de un determinado medicamento y los posibles efectos adversos que pueda tener el mismo, dados por diferentes situaciones, siendo la sobredosificación de éstos la más común o como se evidencia en los antiarrítmicos, la producción paradójica de arritmias dependiente de estos mismos mecanismos de bloqueo de canales iónicos.
Alteración del flujo sanguíneo coronario
El efecto de algunas sustancias se basa en la interacción con el aporte sanguíneo y de oxígeno (O2) al miocardio, con el aumento en los requerimientos de éste y los fenómenos que se asocian a este mecanismo.
Vasoconstricción coronaria
Las sustancias simpaticomiméticas favorecen el aumento del flujo sanguíneo coronario indirectamente mediante los efectos b-adrenérgicos sobre el corazón. Cuando los receptores b se encuentran bloqueados prevalece la acción a-adrenérgica que tienen estas sustancias sobre las arterias coronarias, estimulando la vasoconstricción y por tanto la disminución del flujo sanguíneo al miocardio. Otra consecuencia de la estimulación de los receptores a es la acción simpaticomimética sobre el miocardio previamente afectado, ya que aumenta el cronotropismo e inotropismo y disminuye la respuesta adecuada a la estimulación b, conduciendo finalmente a falla cardíaca. Existen otras sustancias que pueden causar este mismo efecto como la histamina y el tromboxano A2 (2).
Lesión isquemia-reperfusión
Posterior a la lesión isquémica del miocardio, el retorno del aporte sanguíneo al área previamente lesionada puede producir daño temporal conocido como aturdimiento miocárdico, el cual se caracteriza por disfunción contráctil de los miocitos. Este fenómeno se produce por los efectos tóxicos que tienen las sustancias que nuevamente son entregadas al miocito en condiciones de acidosis, disminución de ATP e hipoxia. Para tratar de explicar estos cambios existen diversas teorías, como las paradojas del O2, del pH y del Ca+2. La paradoja del O2 propone la producción de radicales libres posterior al ingreso de O2 (ver más adelante estrés oxidativo). La paradoja del pH propone que la acidosis producida por la isquemia es inicialmente protectora, y el regreso al pH fisiológico, posterior a la reperfusión, elimina esta protección afectando a la célula. La paradoja del Ca+2 propone que la sobrecarga de Ca+2 intracelular posterior a la reperfusión favorece el desarrollo de mecanismos de apoptosis celular (2).
Eliminación del pre-acondicionamiento isquémico
Existen teorías acerca de la posible protección miocárdica del corazón expuesto a isquemia prolongada producida por varios episodios cortos previos de disminución del flujo. Una de las posibles explicaciones está dada por la acción de la adenosina sobre los canales de K+ dependientes de ATP; éstos impiden la sobrecarga de Ca+2 citoplasmático y mitocondrial, ahorran ATP y favorecen la respiración. Por lo tanto, los tóxicos que bloqueen los canales de K+ dependientes de ATP como la glibenclamida, impiden el desarrollo del efecto de pre-acondicionamiento isquémico (2).
Estrés oxidativo
Muchos tóxicos tienen la propiedad de incrementar la producción de radicales libres, también conocidos como especies reactivas de oxígeno, por medio de diferentes fenómenos enzimáticos. Estos radicales son moléculas con electrones desapareados, altamente reactivos, como el anión superóxido, el peróxido de hidrógeno, el radical hidróxilo, el peroxinitrito entre otros, los cuales pueden cambiar las propiedades químicas de una molécula al entrar en contacto con ella; esto se ve reflejado principalmente en la lipoperoxidación de la membrana celular, aumentando su fluidez y la permeabilidad (2). Adicionalmente, el aumento de la concentración de radicales libres en el cardiomiocito activa MAP kinasas (ver más adelante), especialmente la isoforma p38. Las enzimas encargadas de la degradación de radicales libres son la superóxido dismutasa, la catalasa y la glutatión peroxidasa.
Se ha descrito otro mecanismo de daño por estrés oxidativo causado por intoxicación con catecolaminas, donde éstas sufren oxidación espontánea y producen metabolitos oxidativos llamados aminocromos, cuyos efectos incluyen el aumento del flujo de calcio al interior de la célula, que en exceso disminuye la producción de energía y lleva a disfunción contráctil. Ello se asocia con el fenómeno de reciclaje que tiene como resultado la regeneración de catecolaminas oxidables hacia nuevas moléculas de aminocromos y la conjunta producción de nuevos radicales libres (10).
Disfunción de organelos intracelulares
Para el adecuado funcionamiento celular se requiere la integración precisa de todos sus componentes u organelas; por lo tanto, si alguno de estos falla, la célula entra en un estado de disfunción que puede llevar a una alteración funcional total y a la muerte. Dentro de la célula miocárdica, los principales organelos afectados por las sustancias tóxicas son la mitocondria y el retículo sarcoplasmático.
Disfunción mitocondrial
La función principal de la mitocondria es realizar los procesos de respiración celular. La interacción de una sustancia tóxica con este organelo conlleva consecuencias letales para la célula. Las sustancias que intervienen en la respiración celular, actúan a nivel de la fosforilación oxidativa; es el caso de la rotenona, presente en el barbasco, que ejerce su acción a nivel de la unión entre el NADH y la coenzima Q; la actinomicina actúa en la unión entre la coenzima Q y el citocromo C, y el cianuro y el monóxido de carbono lo hacen a nivel del transporte de electrones desde la citocromo oxidasa hasta la molécula de oxígeno (2). El daño mitocondrial puede ocurrir por la liberación del citocromo C desde el espacio intermembranoso hacia el citoplasma, mediado por dos procesos: el primero es la aparición de poros de transición de permeabilidad mitocondrial que provocan tumefacción, ruptura de la membrana externa y liberación del contenido del espacio intermembranoso. El segundo es la sobre-expresión de la proteína Bax que se transloca con el citocromo C a través de la membrana externa. La liberación del citocromo C activa la cascada de apoptosis y, a través de la disminución de ATP por la interrupción de la cadena respiratoria, desencadena a la vez procesos de necrosis celular (3).
Disfunción del retículo sarcoplasmático
Esta organela tiene como función básica la homeostasis del calcio en la célula miocardica; su daño trae como consecuencia la liberación excesiva de este ión al citoplasma. Si este proceso es sostenido se activa la calcineurina que a su vez estimula genes que causan hipertrofia miocárdica (3). La concentración citoplasmática elevada de calcio activa apoptosis celular a través de la estimulación de proteasas y endonucleasas específicas citoplasmáticas, efecto producido por ciertas sustancias como la doxorrubicina, las catecolaminas y la cafeína (2, 5).
Necrosis y apoptosis
Como resultado de los estímulos tóxicos a nivel del cardiomiocito y en especial si son severos, pueden desencadenarse cascadas de procesos intracelulares que median la muerte celular, conocidos como apoptosis y necrosis. Éstos producen pérdida celular global a nivel cardiaco que no se recupera posteriormente y que sumado a los daños causados por otros mecanismos, ya descritos, empeora el cuadro de falla establecido.
La apoptosis, como mecanismo de muerte celular dependiente del consumo de ATP, involucra una cascada de reacciones intracelulares que son el resultado final de diferentes cardiotóxicos; el más común de ellos es la doxorrubicina (ver adelante antineoplásicos), aunque se ha descrito que sólo un pequeño número de células cardiacas muere mediante este mecanismo (2, 3).
La necrosis, a diferencia de la apoptosis, es un modelo de muerte celular independiente de ATP por lo cual, y paralelamente a la apoptosis, puede ser activado por la exposición intensa y prolongada del sistema cardiovascular a una noxa determinada, con la consecuente pérdida de moléculas de ATP intracelulares, mediante consumo energético excesivo o por bloqueo de la cadena respiratoria y la resultante disminución de producción de ATP. Si ésta es mayor al 70% del pool inicial, no se genera suficiente energía para llevar a cabo la apoptosis y se deriva la cascada hacia la necrosis (3).
Sustancias cardiotóxicas
«Todo es veneno, nada es sin veneno. Sólo la dosis hace el veneno» (Paracelso).
Como se mencionó antes, la mayoría de las sustancias con potencial cardiotóxico se utilizan comúnmente como medicamentos benéficos para la salud de los pacientes, pero en dosis o escenarios clínicos inadecuados pueden resultar potencialmente lesivas para el sistema cardiovascular. Por otra parte, existen un sinnúmero de sustancias no medicamentosas que están en contacto con humanos habitualmente y tienen también potencial cardiotóxico. Otras, por su alta toxicidad, han sido controladas estrictamente para disminuir su efecto sobre la población.
A continuación se muestran los efectos tóxicos del alcohol, la cocaína, la clozapina, los antineoplásicos, los anti-inflamatorios no esteroideos y las catecolaminas. Todas estas sustancias vienen siendo ampliamente estudiadas en la actualidad por su potencial toxicidad y el creciente consumo por la población mundial.
Alcohol y cocaína: cocaetileno
Dentro de las sustancias de abuso de mayor consumo en el mundo se encuentran el alcohol y la cocaína, que individualmente tienen efectos cardiotóxicos severos y con su uso concomitante se ven potenciados por numerosas causas, entre ellas la producción de un metabolito activo conocido como cocaetileno.
El consumo frecuente de alcohol en una intensidad leve a moderada, ha demostrado disminuir el riesgo de enfermedad cardiovascular en comparación con los pacientes que no lo consumen o con los que lo hacen con un patrón episódico frecuente, mediante la disminución de la agregabilidad plaquetaria y el mejor control de las cifras de presión para cada una de las comparaciones, respectivamente (11). Con el consumo elevado de alcohol este riesgo aumenta y se evidencian alteraciones, tanto agudas como crónicas.
El etanol tiene efecto dromotrópico negativo y acorta el periodo refractario efectivo auricular, lo que asociado con los efectos sobre la estructura, el tamaño auricular y el estrés oxidativo, demostrados con el consumo exagerado de alcohol, favorece la aparición de fibrilación auricular y otras arritmias (12). Crónicamente, puede causar cardiopatía alcohólica, la cual se caracteriza por alteración de la función sistólica y diastólica del ventrículo izquierdo debido a fibrosis intersticial, disminución del número de células miocárdicas por daño mitocondrial y de la estructura miofibrilar, disminución de la respuesta del aparato contráctil al calcio secundaria a baja concentración de este ión en el retículo sarcoplasmático, disminución de la síntesis proteica e incremento del estrés oxidativo (13, 14); todas estas alteraciones son producidas por el principal metabolito del alcohol, el acetaldehído.
La cocaína, alcaloide considerado en la actualidad como droga de abuso con alto impacto social, tiene efectos cardiotóxicos bien conocidos causados por bloqueo de los canales de sodio (ver atrás mecanismos de arritmogénesis) y bloqueo de receptores a2-presinápticos con inhibición de la recaptación de catecolaminas (15,16). Debido a sus efectos simpaticomiméticos, la cocaína se comporta como inotrópico y cronotrópico positivo, aumenta la tensión arterial y la demanda miocárdica de oxígeno, y promueve fenómenos isquémicos por el efecto vasoconstrictor coronario causado por la actividad agonista a-adrenérgica, debida a su vez a sobrecarga de catecolaminas en la hendidura sináptica, además de la producción de endotelina-1 y la disminución de la producción de óxido nítrico. En el consumidor crónico, estos mecanismos se asocian con el desarrollo de disfunción ventricular sistólica y diastólica, y alteración en la expresión génica del colágeno miocárdico y la miosina. Agudamente se pueden observar alteraciones globales de la función ventricular izquierda, alteraciones focales en la contractilidad debidas a necrosis cardiomiocítica, miocarditis focal (15) y desarrollo del síndrome de Tako-Tsubo (alteración segmentaria ventricular que simula un infarto agudo del miocardio), por aumento de la carga de catecolaminas. El consumo de cocaína favorece el aumento de la agregabilidad plaquetaria (15) y la liberación del inhibidor de activador de plasminógeno, lo que induce la trombosis coronaria (17). Con el abuso de esta sustancia también se asocian la hipertensión y la aceleración del proceso aterosclerótico (16).
El uso combinado de alcohol y cocaína tiene efecto cardiotóxico más severo y mayor mortalidad que el consumo individual de cualquiera de las dos. La potencialización de los efectos se debe a la intensificación de la absorción de la cocaína, la limitación de su excreción y, con ellas, el aumento de la disponibilidad en el organismo, y por la generación de una segunda sustancia denominada cocaetileno, producto de la transesterificación de la cocaína, cuyo metabolismo normal es alterado por la administración previa de etanol (17). Esta sustancia posee varias propiedades similares a las ya descritas para la cocaína, y adicionalmente incrementa con mayor intensidad la frecuencia cardiaca y la demanda miocárdica de oxígeno (18). El cocaetileno es un bloqueador más potente de los canales de sodio y a diferencia de la cocaína, bloquea los canales de calcio ensanchando el QRS y prolongando el segmento QT en el electrocardiograma; así mismo, disminuye la concentración intracitoplasmática de calcio con un efecto inotrópico negativo que se ve enmascarado por el efecto simpaticomimético de la cocaína (19).
Catecolaminas
El sistema nervioso autónomo simpático ejerce varios efectos en el organismo mediante sustancias neurotransmisoras conocidas como catecolaminas, con acción importante a nivel del sistema cardiovascular como se ha mencionado previamente. Mediante la liberación excesiva de catecolaminas endógenas o por la administración exógena de éstas, pueden desencadenarse efectos cardiovasculares, especialmente cuando la dosis sobrepasa la actividad de la catecol-orto-metil-transferasa (COMT) y de la mono-amino-oxidasa (MAO), encargadas de su metabolismo (10).
Los agentes simpaticomiméticos tienen acción inotrópica y cronotrópica positiva. Por el efecto cronotrópico se genera predisposición a la aparición de arritmias ventriculares proporcionales a la intensidad de la alteración en la frecuencia cardiaca.
Estos agentes tienen la capacidad intrínseca de inducir hipertrofia ventricular mediante la estimulación de factores de crecimiento miocárdico, proto oncogenes nucleares y MAP (mitogen-activated protein) kinasas con el aumento de la síntesis proteica (20), en asocio con la respuesta hipertófica inducida por otro de los efectos de la estimulación del sistema simpático: el aumento de la tensión arterial. Estos mecanismos traen consecuencias como el aumento de la demanda de oxígeno por el miocardio, así como de los requerimientos de oxígeno por desacoplamiento de la cadena oxidativa debida a disminución de magnesio, lo cual conduce a hipoxia celular y cambios isquémicos del mismo, acentuados por la aparición de vasoespasmo coronario (ver atrás alteraciones del flujo sanguíneo), con un amplio espectro de manifestaciones que van desde eventos isquémicos silentes hasta infarto agudo del miocardio y síndrome de Tako-Tsubo, caracterizado por cambios electrocardiográficos sugestivos de isquemia, evidencia de hipoquinesia transitoria apical, mínima liberación de marcadores cardiacos y alta concentración de catecolaminas (21, 22).
Otros mecanismos lesivos intrínsecos de los simpaticomiméticos son el incremento de la sobrecarga de calcio intracelular por activación de los canales de Ca+2 tipo L mediado por cAMP dependiente de receptores a, el estrés oxidativo por producción de aminocromos (10) y el daño mitocondrial; cuando estos procesos se suman, se inducen otros de muerte celular, que en exposiciones prolongadas alteran el acoplamiento excitación-contracción que favorece la aparición de arritmias ventriculares (23). Así, afectan la función sistólica ventricular, agravada por la disminución de la respuesta del ventrículo a simpaticomiméticos, debido a la regulación negativa que tienen las catecolaminas sobre los receptores a-adrenérgicos y las cascadas de señalización intracelulares (20, 23).
Antipsicóticos
Dentro de los antipsicóticos más utilizados se encuentra la clozapina, medicamento con efectos cardiotóxicos ya establecidos tales como taquicardia, arritmias, hipotensión, cardiomiopatía, pericarditis y miocarditis, principalmente (24, 25).
Se han formulado diferentes hipótesis para la producción de miocarditis. De hecho, se propone que ésta puede ser producida por reacciones de hipersensibilidad reflejadas por la presencia histopatológica de infiltración perivascular por eosinófilos (24). Se ha descrito la influencia que tiene el bloqueo de receptores muscarínicos M2 colinérgicos y su disfunción por especies reactivas de oxígeno sobre la generación de miocarditis (24). También se ha propuesto como mecanismo el aumento asociado de niveles de catecolaminas y de citoquinas como el factor de crecimiento tumoral (TNF-a) que favorece procesos inflamatorios locales (26).
La miocardiopatía, caracterizada por dilatación ventricular, disfunción contráctil e inicio de falla cardiaca, se genera a partir del daño de los cardiomiocitos por radicales libres; también se ha propuesto su origen a partir de reacciones autoinmunes desencadenadas luego de un episodio de miocarditis, como sucede con la aparición de poliserositis, que incluye pericarditis y derrame pericárdico (24).
Al igual que otros antipsicóticos, la clozapina bloquea los canales de potasio y posee efectos anticolinérgicos que favorecen el prolongamiento del intervalo QT y disminuyen el umbral para la generación de arritmias ventriculares tipo torsión de puntas (24, 27).
Antineoplásicos
Las enfermedades neoplásicas están tomando los primeros lugares de morbimortalidad en el mundo actual, así como la cantidad de tratamientos antineoplásicos crece cada día más. Estos medicamentos son ampliamente conocidos por sus efectos tóxicos en diferentes niveles del organismo. Se han estudiado los efectos que ejercen en el sistema cardiovascular, los cuales limitan ampliamente su uso, entre ellos el desarrollo de falla cardiaca, la generación de arritmias, los procesos isquémicos, el tromboembolismo y la hipertensión arterial. Estos efectos cardiotóxicos tienen diferentes mecanismos y consecuencias dependiendo del tipo de antineoplásico que se trate. En este artículo se mencionan los antineoplásicos comúnmente conocidos por su cardiotoxicidad.
Antraciclinas
Antibióticos antitumorales, como la doxorrubicina y la daunorrubicina, tienen efectos cardiovasculares agudos, subagudos y crónicos. Los efectos agudos son raros y se caracterizan por la presencia de arritmias, miopericarditis y cambios transitorios electrocardiográficos como alteraciones del segmento ST, bajo voltaje del QRS y, en algunos casos, prolongación del intervalo QT; los subagudos se presentan hasta ocho meses posteriores al inicio de la terapia, y los crónicos, que se inician incluso años después, se caracterizan por miocardiopatía con disfunción ventricular izquierda. Se ha mostrado relación entre el daño cardiaco y factores de riesgo como la dosis administrada (dosis máxima acumulada 500 mg/m2), patología cardiaca de base, edad avanzada, radioterapia mediastinal previa y administración concomitante de otros antineoplásicos. Como mecanismos de daño cardiaco, las antraciclinas producen disfunción mitocondrial con posterior depleción de ATP, vacuolización del retículo sarcoplasmático, necrosis, producción de radicales libres mediante el complejo doxorrubicina–hierro (que incrementa la formación de superóxido y de peróxido de hidrógeno) (28) e inhibición del dominio reductor de la sintetasa de óxido nítrico (que además provoca la disminución del mismo óxido nítrico), todo esto exacerbado por la disminución de las enzimas antioxidantes (principalmente de la glutatión peroxidasa), y se asocia con la baja concentración de éstas en las células cardiacas. Además, se ha descrito un aumento de la respuesta inmune local, mediada por la expresión de antígenos posterior al daño de la membrana celular causado por el medicamento (29).
Antimetabolitos
Al igual que el 5-fluorouracilo, tienen mecanismos de daño aún sin aclarar, pero se ha descrito isquemia por vasoespasmo y por trombosis coronaria secundaria a alteraciones de la coagulación (29) y a respuestas autoinmunes; adicionalmente posee efecto cardiotóxico directo mediante la acumulación de citrato intracitoplasmático por bloqueo del ciclo de Krebs y cambios histopatológicos como edema intersticial difuso, vacuolización intracitoplasmática e infiltrados inflamatorios, sugiriendo miocarditis tóxica (30, 31).
Ciclofosfamida
Agente alquilante que favorece la producción de radicales libres mediante su metabolito, la fosforamida. Dentro de los efectos cardiotóxicos que posee se encuentran el daño endotelial capilar con posterior hemorragia intersticial, edema y producción de microémbolos intracapilares coronarios e isquemia (30). Además, se han encontrado alteraciones del ritmo y de la conducción expresadas como taquiarritmias, bloqueos AV y cambios electrocardiográficos como QRS ancho y anormalidades inespecíficas del ST y de la onda T (31).
Anticuerpos monoclonales
Como el trastuzumab, producen disfunción ventricular izquierda, con disminución de la fracción de eyección y falla cardiaca congestiva secundaria a la inhibición del receptor del factor de crecimiento epidérmico de cardiomiocitos (ErbB2), fundamental para el crecimiento y supervivencia de estas células. Su bloqueo lleva a depleción de ATP y disfunción contráctil (30).
Taxoides
Este grupo incluye el paclitaxel y el docetaxel, de los que se han documentado arritmias, isquemia miocárdica, hipertensión arterial y falla cardiaca. Estos efectos se explican por el vehículo utilizado en la fabricación de estos medicamentos, conocido como cremophor EL, el cual genera liberación excesiva de histamina produciendo así aumento de la demanda de oxígeno en el miocardio, vasoconstricción coronaria y prolongación del tiempo de conducción a través del nodo AV y del sistema de conducción His-Purkinje. Adicionalmente, disminuyen el metabolismo de algunas antraciclinas potenciando su toxicidad (30).
Otros efectos cardiotóxicos de los medicamentos antineoplásicos en general incluyen la aparición de hipertensión arterial mediada por inhibición del factor de crecimiento del endotelio vascular (VEGF) disminuyendo la producción de óxido nítrico, activando el sistema renina-angiotensina-aldosterona y aumentando así la resistencia periférica (30). Se ha descrito la aparición de eventos tromboembólicos por medicamentos como el cisplatino, el cual favorece la activación y la agregación plaquetaria, altera el endotelio vascular e incrementa el factor de Von Willebrand de la cascada de coagulación, con lo cual, sumado a las alteraciones renales que provoca, se evidencian desequilibrios hidroelectrolíticos que favorecen la aparición de isquemia, arritmias y vasoespasmo (30, 31).
AINE - Inhibidores de la COX-2
Dentro del grupo de antiinflamatorios no esteroideos (AINE) se encuentran aquellos que se caracterizan por inhibir selectivamente la isoenzima ciclooxigenasa-2 (COX-2); éstos no poseen las propiedades benéficas sobre el sistema cardiovascular tan ampliamente divulgadas del ácido acetilsalicílico (ASA), un inhibidor no selectivo de la COX, entre las que se encuentra principalmente la anti-agregación plaquetaria. Estos medicamentos inhibidores selectivos de la COX-2, entre ellos el rofecoxib, el valdecoxib y el celecoxib (único aprobado por la FDA en la actualidad), poseen acción cardiotóxica asociada a su mecanismo de acción pues favorecen la síntesis de tromboxano A2 (TXA2) propiciando un estado protrombótico exacerbado por la inhibición de la prostaglandina I2 (PGI2), que en condiciones normales inhibe la agregación plaquetaria y produce vasodilatación. Entre los efectos cardiotóxicos de estos medicamentos se encuentran los eventos isquémicos miocárdicos; también se han descrito casos de hipertensión arterial por daño de la función endotelial y de la función renal producido, como ya se explicó, por la inhibición de la acción vasodilatadora de la PGI2, que lleva al desarrollo de falla cardiaca congestiva y arritmias (32-34).
Aunque se ha establecido el riesgo de eventos cardiovasculares con el uso de inhibidores de la COX-2, este efecto varía entre los diferentes integrantes de este grupo, siendo el rofecoxib el más estudiado. Cabe resaltar que dicha toxicidad también depende de la dosis y la duración del tratamiento. Al día de hoy no existen estudios que muestren algún tipo de protección con el uso concomitante de ASA (33, 35).
Se debe aclarar que no todos los inhibidores no selectivos de la COX tienen efecto cardioprotector como el ASA. Con el uso de diclofenaco se ha evidenciado aumento del riesgo cardiovascular a largo plazo; utilizar ibuprofeno en forma concomitante con ASA disminuye su efecto antiagregante plaquetario debido a que compiten por su sitio de unión a la COX-1 (35).
Conclusiones
A lo largo de los años se han descrito numerosos mecanismos de daño cardiaco, que han permitido entender las alteraciones en el sistema cardiovascular que se observan durante o luego de la exposición a determinados fármacos o sustancias. Esto, a su vez, condujo a la creación de ciertos límites para su uso, incluso su salida definitiva del mercado por la severidad de sus efectos. Dentro de los mecanismos y consecuencias de cardiotoxicidad se encuentran la producción de alteraciones en el ritmo y la frecuencia cardiaca mediante el bloqueo de canales iónicos de membrana y estímulo autonómico, el daño a organelos intracelulares por disfunción de proteínas transportadoras, la producción de radicales libres que desestabilizan o alteran las cadenas de señalización intracelular conduciendo a procesos de muerte celular tipo necrosis o apoptosis, así como procesos de compensación que a largo plazo se traducirán en mal funcionamiento cardiaco.
Entre las sustancias que han sido estudiadas desde tiempo atrás y que son ya conocidas por su toxicidad cardiaca, se encuentra el antipsicótico clozapina y las catecolaminas, que favorecen el desarrollo de falla cardiaca, procesos isquémicos y arritmias. Estos estudios han llevado a la búsqueda de noxas cardiacas en sustancias de creciente uso en la actualidad, como los AINE, que han demostrado tener efectos protrombóticos desencadenantes de eventos isquémicos, y los antineoplásicos, que abarcan una gran cantidad de fármacos que lesionan de manera aguda, subaguda y crónica el miocardio por medio del estrés oxidativo, la disfunción mitocondrial y el retículo sarcoplasmático, y acarrean el desarrollo de mecanismos compensadores mal-adaptativos. Así mismo, se han estudiado los efectos del consumo de sustancias de abuso como el alcohol y la cocaína y las consecuencias de su uso concomitante con la producción de cocaetileno, que potencia los daños que pueden ser causados por ambas sustancias de manera independiente.
Como consideración final cabe resaltar la importancia de esta revisión como base teórica para futuras investigaciones y profundizaciones al respecto, así como la consecuente aplicación clínica para el adecuado manejo del paciente intoxicado.
Bibliografía
1. Neumar RW, Otto CW, Link MS, et al. Part 8: adult advanced cardiovascular life support: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation 2010; 122 (18) (Suppl 3): S729-67. [ Links ]
2. Ramos KS, Melchert RB, Chacón E, Acosta D. Toxic responses of the heart and vascular systems. En: Casarett LJ, Doull J, Klaassen CD. Casarett and Doull's Toxicology: The Basic Science of Poisons. 6th ED. New York: McGraw Hill, 2001. p. 597-631. [ Links ]
3. Kang YJ. Molecular and cellular mechanisms of cardiotoxicity. Environ Health Perspect 2001; 109 (suppl 1): 27-34 [ Links ]
4. Bohórquez LF, Pineda M, Marín F, et al. Bases Fundamentales de la Cardiología. En: Acuña J, Anchique CV, Arango JJ, et al. Texto de Cardiología. Sociedad Colombiana de Cardiología y Cirugía Cardiovascular. Bogotá D.C., Colombia: Legis S.A.; 2007. p. 3-81. [ Links ]
5. Eisner DA, Kashimura T, O'Neil SC, Venetucci, Trafford AW. What role does modulation of the ryanodine receptor play in cardiac inotropy and arrhythmogenesis? J Mol Cell Cardiol 2009; 46 (4): 474-81. [ Links ]
6. Hessler RA. Cardiovascular principles. En: Flomenbaum N, Goldfrank L, Hoffman R, Howland MA, Lewin N, Nelson, L. Goldfrank's Toxicologic Emergencies. 8th ED. New York: McGraw Hill; 2006. p. 364-77. [ Links ]
7. Patel MM, Benowitz N. Cardiac conduction and rate disturbances. En: Brent J. Critical care toxicology: diagnosis and management of the critically poisoned patient. St. Louis: Elsevier Mosby; 2005. p. 241-57. [ Links ]
8. Delk C, Holstege CP, Brady WJ. Electrocardiographic abnormalities associated with poisoning. Am J Emerg Med 2007; 25 (6): 672-87. [ Links ]
9. Vaughan Williams EM. Significance of classifying antiarrhythmic actions since the cardiac arrhythmia suppression trial. J Clin Pharmacol 1991; 31 (2): 123-35. [ Links ]
10. Behonick GS, Novak MJ, Nealley EW, Baskin SI. Toxicology update: the cardiotoxicity of the oxidative stress metabolites of catecholamines (aminochromes). J Appl Toxicol 2001; 21 (Suppl 1): S15-22. [ Links ]
11. Mukamal KJ, Conigrave KM, Mittleman MA, et al. Roles of drinking pattern and type of alcohol consumed in coronary heart disease in men. N Engl J Med 2003; 348 (2): 109-18. [ Links ]
12. Mukamal KJ, Tolstrup JS, Friberg J, Jensen G, Grønbæk M. Alcohol consumption and risk of atrial fibrillation in men and women: the Copenhagen City Heart Study. Circulation 2005; 112 (12): 1736-42. [ Links ]
13. Liu J, Yano M, Shimamoto A, Noma T, Matsuzaki M, Fujimiya T. Chronic effects of ethanol on pharmacokinetics and left ventricular systolic function in rats. Alcohol Clin Exp Res 2007; 31 (3): 493-9. [ Links ]
14. Kennedy RH, Liu SJ. Sex differences in L-type calcium current after chronic ethanol consumption in rats. Toxicol Appl Pharmacol 2003; 189 (3): 196-203. [ Links ]
15. Kloner RA, Hale S, Alker K, Rezkalla S. The effects of acute and chronic cocaine use on the heart. Circulation 1992; 85 (2): 407-19. [ Links ]
16. Afonso L, Mohammad T, Thatai D. Crack whips the heart: a review of the cardiovascular toxicity of cocaine. Am J Cardiol 2007; 100 (6): 1040-3. [ Links ]
17. Harris DS, Everhart ET, Mendelson J, Jones RT. The pharmacology of cocaethylene in humans following cocaine and ethanol administration. Drug Alcohol Depend 2003; 72 (2): 169-82. [ Links ]
18. Pennings EJ, Leccese AP, de Wolff FA. Effects of concurrent use of alcohol and cocaine. Addiction 2002; 97 (7): 773-83. [ Links ]
19. Wilson LD, Jeromin J, Garvey L, Dorbant A. Cocaine, ethanol, and cocaethylene cardiotoxity in an animal model of cocaine and ethanol abuse. Acad Emerg Med 2001; 8 (3): 211-22. [ Links ]
20. Osadchii OE. Cardiac hypertrophy induced by sustained beta-adrenoreceptor activation: pathophysiological aspects. Heart Fail Rev 2007; 12 (1): 66-86. [ Links ]
21. Pernicova I, Garg S, Bourantas CV, Alamgir F, Hoye A. Tako-Tsubo cardiomyopathy: a review of the literature. Angiology 2009; 61 (2): 166-73. [ Links ]
22. Vidi V, Rajesh V, Singh PP, et al. Clinical characteristics of Tako-Tsubo cardiomyopathy. Am J Cardiol 2009; 104 (4): 578-82 [ Links ]
23. Feldman DS, Elton TS, Sun B, Martin MM, Ziolo MT. Mechanisms of disease: detrimental adrenergic signaling in acute decompensated heart failure. Nat Clin Pract Cardiovasc Med 2008; 5 (4): 208-18. [ Links ]
24. Merrill DB, William GW, Goff DC. Adverse cardiac effects associated with clozapine. J Clin Psychopharmacol 2005; 25 (1): 32-41. [ Links ]
25. Wehmeier PM, Heiser P, Remschmidt H. Myocarditis, pericarditis and cardiomyopathy in patients treated with clozapine. J Clin Pharm Ther 2005; 30 (1): 91-6. [ Links ]
26. Wang JF, Min JY, Hampton TG, et al. Clozapine-induced myocarditis: role of catecholamines in a murine model. Eur J Pharmacol 2008; 592 (1-3): 123-7. [ Links ]
27. Harrigan EP, Miceli JJ, Anziano R, et al. A randomized evaluation of the effects of six antipsychotic agents on QTc, in the absence and presence of metabolic inhibition. J Clin Psychopharmacol 2004; 24 (1): 62-9. [ Links ]
28. Simùnek T, Stérba M, Popelová O, Adamcová M, Hrdina R, Gersl V. Anthracycline-induced cardiotoxicity: overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol Rep 2009; 61 ( 1): 154-71. [ Links ]
29. Schimmel KJ, Richel DJ, van den Brink RB, Guchelaar HJ. Cardiotoxicity of cytotoxic drugs. Cancer Treat Rev 2004; 30 (2): 181-91. [ Links ]
30. Yeh ET, Bickford CL Cardiovascular complications of cancer therapy: incidence, pathogenesis, diagnosis, and management. J Am Coll Cardiol 2009; 53 (24): 2231-47. [ Links ]
31. Floyd JD, Nguyen DT, Lobins RL, Bashir Q, Doll DC, Perry MC. Cardiotoxicity of cancer therapy. J Clin Oncol 2005; 23 (30): 7685-96. [ Links ]
32. Brophy, JM. Cardiovascular effects of cyclooxygenase-2 inhibitors. Curr Opin Gastroenterol 2007; 23 (6): 617-24. [ Links ]
33. Mukherjee D. Nonsteroidal anti-inflammatory drugs and the heart: what is the danger? Congest Heart Fail 2008; 14 (2): 75-82. [ Links ]
34. Patrono C, Baigent C, Hirsh J, Roth G. American College of Chest Physicians. Antiplatelet drugs: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th. Edition). Chest 2008; 133 (Suppl 6): 199S-233S. [ Links ]
35. Farkouh ME, Greenberg BP. An evidence-based review of the cardiovascular risks of nonsteroidal anti-inflammatory drugs. Am J Cardiol 2009; 103 (9): 1227-37. [ Links ]

