










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Cardiología
Print version ISSN 0120-5633
Rev. Colomb. Cardiol. vol.19 no.1 Bogota Jan./Feb. 2012
Amiloidosis cardiaca: presentación de un caso y revisión de la literatura
Cardiac amyloidosis: a case report and review of literature
Luis Enrique Núñez Moscoso, MD. (1); Sharlot Chacón Arévalo, MD.(1)
Institución en la cual se realizó el trabajo: Servicio de Medicina Interna. Hospital I Edmundo Escomel –EsSalud. Arequipa, Perú. Av. El Cayro s/n –Paucarpata
(1) Hospital I Edmundo Escomel –EsSalud. Arequipa, Perú.
Correspondencia: Dr. Luis Enrique Núñez Moscoso. Hospital I Edmundo Escomel. Arequipa. Av. El Cayro s/n Paucarpata. Teléfono: (51) 054 38 04 30 Ext.: 302. Correo electrónico: lnunez10@hotmail.com
Recibido: 06/12/2010. Aceptado: 25/05/2011.
Usualmente, el daño cardiaco en la amiloidosis poco se reconoce debido a su presentación inespecífica y al diagnóstico tardío, lo cual puede llevar a errores tanto en el tratamiento como en la información que se comunica al paciente. La excepcional presentación y la afectación variable de diferentes órganos y tejidos hacen de esta entidad un reto clínico considerable.
Se presenta el caso de un paciente de género masculino, con signos de disfunción cardiaca diastólica severa y se analizan los hallazgos de la historia clínica y los datos de ayuda al diagnóstico. Finalmente, se hace una revisión de la literatura.
Palabras clave: amiloidosis cardiaca, cardiomiopatía restrictiva, amiloidosis primaria.
Cardiac damage in amyloidosis is usually little recognized due to its non specific presentation and delayed diagnosis, which can lead to errors both in treatment and information given to the patient. The exceptional presentation and variable involvement of different organs and tissues make this entity a significant clinic challenge.
We present the case of a male patient with signs of severe diastolic cardiac disfunction and analyze the findings of the medical record and diagnostic data. Finally, we make a literature review.
Key words: cardiac amyloidosis, restrictive cardiomyopathy, primary amyloidosis.
Paciente varón, de 49 años de edad, sin antecedentes personales ni familiares de importancia, que refiere un tiempo de enfermedad de cuatro meses, de inicio insidioso y curso progresivo con la aparición de mareos, debilidad generalizada y visión borrosa al deambular. A ello se le añade que un mes antes del ingreso presentó aumento de volumen de miembros inferiores de tipo vespertino, disnea de esfuerzo, dolor torácico y distensión abdominal. Al examen físico de ingreso se le halló despierto, lúcido, colaborador con la evaluación, presión arterial de 90/50 mm Hg, frecuencia cardiaca de 92 latidos por minuto, ruidos cardiacos regulares sin presencia de soplos, ingurgitación yugular II/IV, ortopnea, pulmones con murmullo vesicular abolido en ambas bases a predominio derecho, matidez en ambas bases, abdomen globuloso, blando con matidez desplazable, hígado palpable a 7 cm por debajo del reborde costal derecho, bordes romos dolorosos, no se palpó bazo, edema de miembros inferiores hasta pubis III/III.
Adicionalmente, se realizaron las siguientes pruebas de laboratorio: hemograma: leucocitos 12 670/mm3, 45% neutrófilos, 39% linfocitos, 10% monocitos, 3% eosinófilos, 3% basófilos; hemoglobina 15 g/dL; hematocrito 46,4 mL/dL, plaquetas 201 000/ mm3, glucosa 70 mg/dL, creatinina 1,34 mg/dL, DHL 442 UI, fosfatasa alcalina 997 mU/mL, TGO(ASAT) 45 mU/mL, TGP(ALAT) 47 mU/mL, GGTP 925 UI, globulina 2,74 g/dL, albúmina 3,41 g/dL, bilirrubinas totales 1,54 mg/dL, bilirrubinas indirectas 0,84 mg/dL, bilirrubinas directas 0,7 mg/dL, VSG 14 mm/H, PCR 42,4 mg/dL, depuración de creatinina 46,36 mL/min, proteinuria de 24 horas 1,4 g/L, C3 121 mg/dL, C4 53,8 mg/dL; sedimento urinario: leucocitos 3 a 4 por campo, hematíes 0 a 2 por campo, bacterias +. BK en esputo I; II; III negativo.
Así mismo, se realizaron estudios radiográficos (Figura 1), tomográficos (Figura 2) y ecocardiográficos (Figuras 3, 4, 5), y un procedimiento diagnóstico (Figura 6).
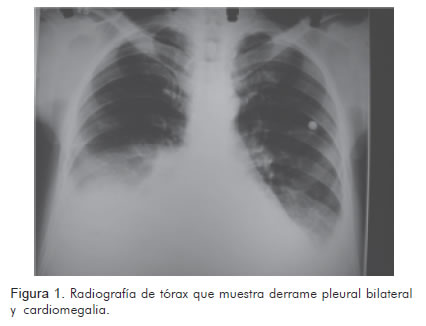
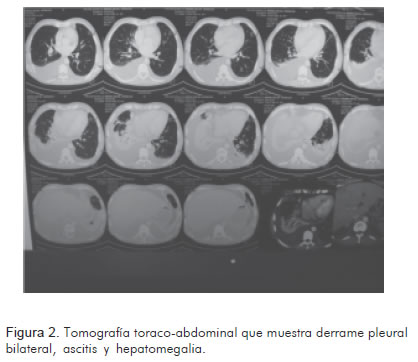
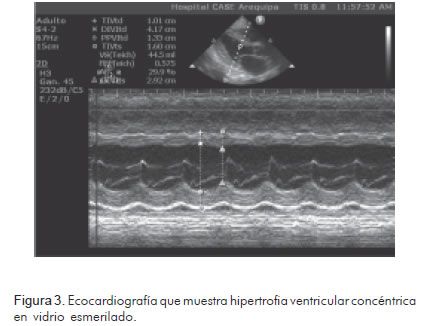
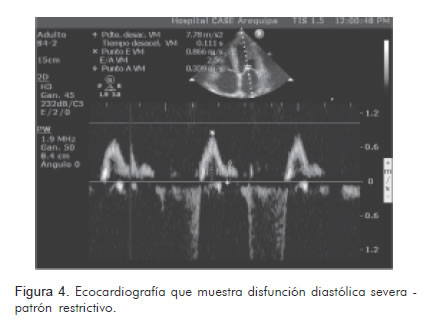

Durante su hospitalización presentó una evolución desfavorable, con mala respuesta al tratamiento instaurado, y falleció de forma súbita luego de quince días de internamiento.
DiscusiónLa amiloidosis se conoce desde hace más de 300 años. El término «amiloide» fue acuñado en 1838 por Mathias Schleiden, botánico alemán, para describir un constituyente normal de las plantas. En 1854, Rudolf Vichow observó algunas propiedades tincionales similares al amiloide en autopsias de hígado (1). En la actualidad se considera no como una sola enfermedad sino como un conjunto de enfermedades que comparten un hecho común: el depósito extracelular de proteínas fibrilares insolubles patológicas en órganos y tejidos (2).
La sustancia amiloidea parece ser homogénea y amorfa a la microscopia de luz; las tinciones metacromáticas, como el violeta de metilo o el violeta de cristal, producen típicos cambios de coloración en el tejido amiloide. La tinción con rojo congo se considera más específica; cuando es visualizada con una fuente de luz polarizada produce una birrefringencia verde manzana. Uno de los descubrimientos principales fue el reconocimiento de que las fibrillas amiloideas en la amiloidosis primaria eran fragmentos de las cadenas ligeras de las inmunoglobulinas (3). Subsecuentemente se determinó que diferentes proteínas formaban parte de las fibrillas amiloideas en las amiloidosis secundarias y familiares (Tabla 1) (4).
El mecanismo fisiopatológico por el cual el depósito de amiloide causa daño en los tejidos, no es del todo conocido. Estudios recientes demuestran que existen más de veinte proteínas que pueden formar depósitos fibrilares extracelulares. El amiloide no está compuesto sólo por fibrillas sino por proteoglicanos, componente P y factor liberador de amiloide. Este último parece ser el punto precipitante de la activación celular, la formación amiloidea y el daño subsiguiente (5).
La epidemiología de la amiloidosis es difícil de precisar. Se estima una incidencia ajustada por edad y género de entre 5,1 y 12,8 /millón/persona–año, lo que significa aproximadamente entre 1.275 y 3.200 nuevos casos al año, en los Estados Unidos (6). No existen datos registrados en Latinoamérica y especialmente en Perú ya que la mayor cantidad de registros corresponde a revisiones de casos y estudios pos-mortem (7).
La afectación cardiaca en la amiloidosis primaria es de relativa frecuencia ya que en algunas series llega casi al 50% de casos y la falla cardiaca congestiva es la manifestación clínica inicial en cerca de la mitad de estos pacientes. A pesar de que haya disfunción predominante en otros órganos, la presencia la amiloidosis cardiaca es, con frecuencia, el peor factor pronóstico. Una vez instalada la falla cardiaca el tiempo promedio de supervivencia es menor de seis meses en pacientes no tratados. El caso típico con falla cardiaca resultante de amiloidosis, generalmente se presenta con síntomas y signos de progreso vertiginoso. La disnea progresiva es común generalmente se asocia con presión elevada de llenado del ventrículo derecho. El edema periférico puede ser profundo y en estados avanzados de la enfermedad, no es infrecuente la ascitis. El examen físico cardiovascular revela ritmo sinusal con volumen de pulso radial normal o bajo. De igual manera, algunas formas de arritmias auriculares ocurren en 10% a 15% de pacientes. El pulso venoso yugular está marcadamente elevado. En raras ocasiones se presenta el signo de Kussmaul. Usualmente el primero y segundo ruidos cardiacos son normales, y pocas veces se escucha un tercer ruido. Es común encontrar baja la presión sanguínea, incluso en ausencia de hipotensión postural; esto puede representar un gasto cardiaco disminuido junto con disfunción autonómica temprana (6).
El electrocardiograma muestra bajo voltaje en una gran proporción de pacientes, aunque puede observarse un patrón de pseudo-infarto el cual puede ser mal interpretado como isquemia silente. Los bloqueos de rama, sobre todo derecha, se presentan de forma inusual (7).
Los patrones ecocardiográficos en la amiloidosis cardiaca avanzada son distintivos y se describen como en «vidrio esmerilado». La aparición clásica de un patrón restrictivo asociado al incremento de la ecogenicidad, el engrosamiento biventricular y la infiltración valvular, está limitada a pacientes en el estadio final de la enfermedad. La fracción de eyección del ventrículo izquierdo es normal o cercana a lo normal. Además, existe una correlación entre los hallazgos ecocardiográficos y el pronóstico de los pacientes (8). De esta forma, quienes tienen un grosor de las paredes septal y posterior menor o igual a 12 mm tienen un promedio de vida de 2,4 años y habitualmente no sufren disfunción sistólica. Quienes tienen un grosor entre 12 a15 mm presentan un promedio de vida de 1,3 años y 35% de ellos presenta disfunción sistólica. Finalmente, aquellos con un grosor de las paredes mayor o igual a 15 mm, tienen un promedio de vida de 0,4 años y 70% de ellos tienen disfunción sistólica. Así, existe una clara relación entre el deterioro clínico y la progresión del engrosamiento de las paredes del ventrículo izquierdo; sin embargo, la dilatación ventricular sólo se aprecia en 5% de los pacientes (9, 10).
El diagnóstico de amiloidosis requiere la demostración histopatológica de los depósitos de amiloide. La aspiración de grasa abdominal a través de aguja fina tiene una sensibilidad de 87%, es inocua y puede sustituir a la biopsia de los órganos involucrados. Una vez que se establece el diagnóstico histopatológico, se define el tipo de amiloide (6, 11, 12). La caracterización de una amiloidosis confirmada por biopsia como tipo AL requiere la demostración de la activación de una clona de células plasmáticas subyacente. Se recomienda hacer esta demostración por electroforesis de inmunofijación tanto en suero como de orina, la cual puede detectar una proteína monoclonal hasta en 97% de los casos.
El Décimo Simposio Internacional sobre amiloide y amiloidosis define la afectación de órgano blanco (Tabla 2), así como los criterios para evaluación de la respuesta al tratamiento y la progresión de la enfermedad (10).
El tratamiento de la amiloidosis cardiaca requiere una aproximación desde dos puntos: el manejo de los síntomas cardíacos y el tratamiento de la enfermedad de fondo (8). Uno de los pilares es el uso de diuréticos. Los inhibidores de la enzima convertidora de angiotensina y los ARA II son pobremente tolerados; dosis pequeñas pueden producir hipotensión profunda y no hay datos de la efectividad de los beta-bloqueadores en la mejoría de la sobrevida en esta condición. De otra parte, los bloqueadores de los canales de calcio están contraindicados debido a su efecto inotrópico negativo. El uso de digoxina es de poco valor y se corre el riesgo de toxicidad digitálica debido a que este medicamento se une ávidamente a las fibrillas de amiloide. El tratamiento definitivo es la terapia contra las células plasmáticas productoras de la paraproteína. Existe un variado número de regímenes quimioterapéuticos. Infortunadamente, la naturaleza avanzada de la enfermedad cardiaca en muchos pacientes al momento del diagnóstico contraindica el tratamiento tanto quimioterapéutico como el reemplazo con células madre autólogas; además, la mortalidad peritratamiento puede ser de hasta 30%. Finalmente, en pacientes seleccionados puede considerarse el transplante cardíaco (13).
En conclusión, aunque la amiloidosis cardiaca es poco común, se caracteriza por hallazgos típicos en la ecocardiografía. Debe sospecharse en todo paciente con miocardiopatía restrictiva, ya que éstos desarrollan disfunción diastólica temprana y anomalías no específicas en el sistema de conducción. El diagnóstico temprano es crítico ya que en estados avanzados estos pacientes están muy enfermos para soportar la quimioterapia intensiva. Las modalidades terapéuticas disponibles son sólo medidas de soporte.
Bibliografía1. Kyle R, Gertz M. Primary systemic amyloidosis. Clinical and Laboratory Features in 474 Cases. Seminars in Haematology 1995; 32 (1): 45-59. [ Links ]
2. Bohne S, Sletten K, Menard R et al. Cleavage of Al amyloid protein and AL amyloid deposit by chatepsin B, an L. Pathol 2004; 203: 528-537. [ Links ]
3. Glenner GG, TerryW, Harada M, Iserky C, Page D. Amyloid fibrilm proteins: proof of homology with immunoglobulin light chains by sequence analyses. Science 1971; 172: 1150-1. [ Links ]
4. Kyle R, Gertz M. Amyloidosis. Chapter 3. Systemic diseases and the kidney. 2005. [ Links ]
5. Magy N. AA Amyloidosis: recent knowledges and pathophysiology. Rev Med Int 2004; 25 (2): 129-34. [ Links ]
6. Falk R, Comenzo R, Skinner M. The systemic amyloidosis. N Engl J Med 1997; 337 (13): 898-908. [ Links ]
7. Palma CL, Grunholz D, Osorio G. Amiloidosis. Comunicación de 11 casos y revisión de la literatura. Rev Med Chile 2005; 133: 655-661. [ Links ]
8. Falk R. Diagnosis and management of the cardiac amyloidosis. Circulation 2005; 112: 2047-2060. [ Links ]
9. Rovegno M, Ibáñez R. Caso clínico: amiloidosis cardiaca. Clínica y Ciencia 2001; 1 (2): 35-39. [ Links ]
10. Gertz M, Comenzo R, Falk R et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th. International Symposium on Amyloid and Amyloidosis. Am J Hematol 2005; 79: 319-328. [ Links ]
11. Gertz M, Lacy M, Dispenzerie A, Hayman S. Amyloidosis. Best practice and research clinical haematology 2005; 18 (4): 709-727. [ Links ]
12. Palladini G, Perfetti V, Merlini G. Therapy and management of systemic AL (primary) amyloidosis. Swiss Med Wkly 2006; 136: 715-720. [ Links ]
13. Housenpud JD, Uretsky BF, Griffith BP et al. Successful intermediate-term outcome for patients with cardiac amyloidosis undergoing heart transplantation: result of a multicenter survey. J Heart Transplant 1990; 9: 346-350. [ Links ]

