










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Cardiología
Print version ISSN 0120-5633
Rev. Colomb. Cardiol. vol.20 no.5 Bogota Sept./Oct. 2013
Unidad de Dolor Torácico, Hospital Universitario San Vicente de Paúl. Medellín, Colombia.
(1) Hospital San Vicente de Paúl. Medellín, Colombia.
(2) Clínica Medellín. Medellín, Colombia.
(3) Universidad de Antioquia. Medellín, Colombia.
Correspondencia: Dr. Juan M. Senior, Hospital Universitario San Vicente de Paúl-Universidad de Antioquia. Calle 64 Carrera 51D. Medellín, Colombia. Teléfono: (57-4) 516 7403. Correo electrónico: mmbt@une.net.co
Recibido: 28/11/2011. Aceptado: 11/06/2013.
Paciente de 65 años, con historia de epistaxis a repetición, sin otros antecedentes patológicos de importancia, quien presentó síntomas de un año de evolución consistentes en disnea de esfuerzo progresiva hasta el reposo, acompañado de ortopnea y edemas que iniciaron en miembros inferiores y progresaron hasta la anasarca. Al examen clínico llamó la atención ingurgitación yugular, hepatomegalia, ascitis y edemas periféricos. La piel presentó telangiectasias en extremidades y mucosas. Se realizaron estudios que reportaron falla cardiaca con severa dilatación de las cavidades derechas, con función sistólica del ventrículo izquierdo conservada. Se demostraron fístulas arteriovenosas en hígado y pulmón. Se estableció el diagnóstico de síndrome de Osler-Weber-Rendu o telangiectasia hemorrágica hereditaria (THH). Se propuso cierre de fístulas por vía percutánea y se planteó posible trasplante hepático como tratamiento; sin embargo, el paciente tuvo una evolución tórpida, presentó fibrilación auricular y embolismo al sistema nervioso central y falleció por enfermedad cerebrovascular.
Palabras clave: fístula, insuficiencia cardiaca, cortocircuito, gasto, cateterismo cardiaco.
We report the case of a 65 year old patient with history of recurrent epistaxis without other significant medical history, who presented symptoms of progressive dyspnea, from exertional dyspnea to dyspnea at rest, orthopnea and edema that began in lower limbs and progressed to anasarca. Clinical examination showed jugular ingurgitation, hepatomegaly, ascites and peripheral edema. Skin telangiectasiae were found in extremities and mucosae. Studies performed reported heart failure with severe dilatation of the right cavities with left ventricular systolic function preserved. Arteriovenous fistulas were demonstrated in liver and lung. A diagnosis of Osler-Weber-Rendu syndrome or hereditary hemorrhagic telangiectasia (HHT)was established. Percutaneous closure of fistulas was proposed and a possible liver transplantation was considered as a treatment, but the patient had a torpid evolution, presented atrial fibrillation and embolism to the central nervous system and died from cerebrovascular disease.
Keywords: fistula, heart failure, short, spending cardiac catheterization.
Introducción
La telangiectasia hemorrágica hereditaria (THH) o enfermedad de Osler-Weber-Rendu, es un transtorno genético con dominancia autosómica y penetrancia variable. Característicamente los pacientes presentan epistaxis recurrentes, telangiectasias mucocutáneas y fístulas arteriovenosas viscerales, las cuales pueden desarrollarse en cerebro (9,7%), pulmones (34%), hígado (57%) y tracto gastrointestinal (80%) (1). Su prevalencia se estima en 1:5.000-10.000 habitantes. Es una entidad poco reconocida, cuyo diagnóstico se basa en los hallazgos clínicos; los pacientes pueden fallecer por hemorragia intracerebral o sangrado gastrointestinal masivo (1). Se describe el caso de un paciente en la séptima década de la vida con falla cardiaca de alto débito y complicaciones tardías de la enfermedad.
Presentación del caso
Paciente de 65 años de edad, con historia de epistaxis recurrente, sin otros antecedentes personales ni familiares patológicos de importancia, quien consultó por disnea y edemas que iniciaron un año antes, que en pocos meses progresaron hasta disnea de reposo y anasarca, asociado a ortopnea, pero sin disnea paroxística nocturna.
Al examen clínico se encontró pálido, caquéctico, con presión arterial de 98/64 mm Hg, frecuencia cardiaca de 98 latidos por minuto, frecuencia respiratoria de 24 por minuto y temperatura de 36,7ºC. Saturación de oxígeno con fracción inspirada de oxígeno de 21% de 92%, ingurgitación yugular a 45º. Los ruidos cardiacos eran rítmicos con soplo holosistólico epigástrico II/IV, sin frote pericárdico, sin S3. El punto de máximo impulso, se halló en la región subxifoidea. Los ruidos respiratorios estaban conservados, sin sobreagregados. La palpación del abdomen reveló hepatomegalia severa, sin esplenomegalia, con peristaltismo normal y matidez desplazable presente. Las extremidades tenían edemas con fóvea hasta el muslo. También había edema de la pared abdominal y en la región sacra. En la piel llamaron la atención lesiones eritematosas puntiformes en los pulpejos de los dedos, e igualmente en los labios y en la mucosa oral (Figura 1).
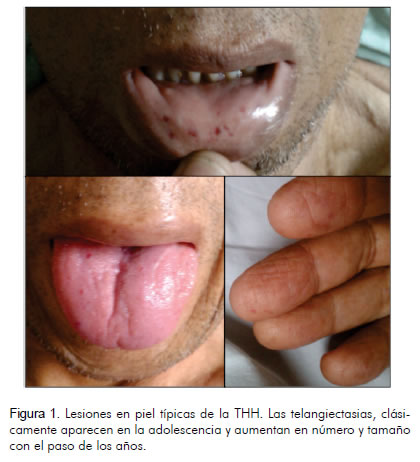
Ingresó al servicio de hospitalización con diagnóstico de falla cardiaca derecha de etiología no clara. Los exámenes paraclínicos demostraron: hemograma con 4.600 leucocitos x mL, polimorfonucleares neutrófilos 3.600 x mL, hemoglobina de 8,6 g/dL, volumen corpuscular medio 88 µm3, hierro 14 mcg/dL, ferritina 81 ng/mL, saturación de transferrina 3,9%, bilirrubina total 2,7 mg/dL, bilirrubina directa 2,1 mg/dL, fosfatasas alcalinas 236 UI/L, creatinina 3,5 mg/dL, nitrógeno ureico 75 mg/dL, depuración de creatinina 18,8 mL/min, TSH 3,2 mLU/L, gases arteriales: pH 7,24, pCO2 28 mm Hg, bicarbonato 18 meq/litro, potasio 5,5 meq/L, sodio 132 meq/L, cloro 99 meq/L, magnesio 2,5 meq/L y tiempo de protrombina 14,9 segundos.
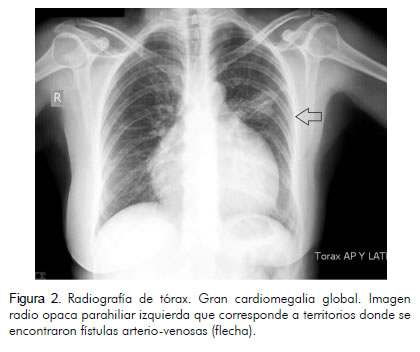
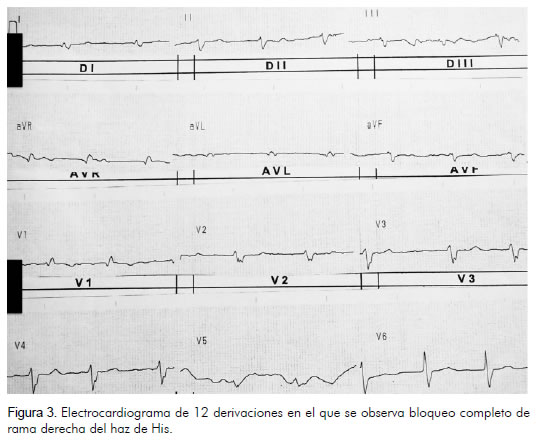
Se realizaron los siguientes estudios de imagen: radiografía de tórax que mostró gran aumento del índice cardiotorácico, radiopacidad parahiliar izquierda correspondiente al territorio donde se demostraron fístulas arteriovenosas (Figura 2), electrocardiograma que mostró bloqueo completo de la rama derecha del haz de His y fibrilación auricular (Figura 3), ecocardiografía con fracción de expulsión del ventrículo izquierdo de 61%, con diámetro diastólico del ventrículo izquierdo de 6 cm y sistólico de 4 cm. El ventrículo derecho estaba severamente dilatado (6 cm). El área de la aurícula izquierda era de 40 cm2 y el de la derecha de 80 cm2. Además, insuficiencia tricuspídea y presión sistólica de la arteria pulmonar estimada en 28 mm Hg (Figura 4).
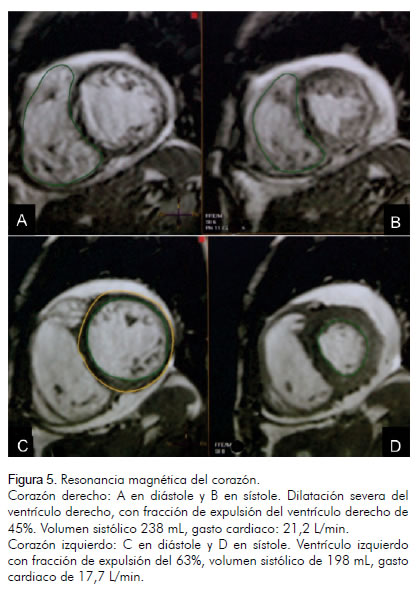
Debido a las lesiones en la piel, se sospechó síndrome de Osler-Weber-Rendu, y se decidió buscar fístulas arteriovenosas sistémicas, por lo cual se realizaron angiotomografías de tórax y de abdomen que mostraron múltiples malformaciones arteriovenosas en el pulmón y en el hígado, sin alteraciones óseas evidentes. La ecografía-doppler del hígado evidenció vena porta de 2 cm y vena cava inferior de 4,1 cm y confirmó las múltiples malformaciones arteriovenosas hepáticas. El estudio cardiovascular se complementó con una resonancia cardiaca (Figura 5) y un cateterismo derecho. La resonancia cardiaca reportó dilatación severa del ventrículo derecho con fracción de expulsión del 45%, volumen sistólico 238 mL, gasto cardiaco 21,2 L/min y ventrículo izquierdo con fracción de expulsión del 63%, volumen sistólico de 198 mL y gasto cardiaco de 17,7 L/min. En el cateterismo derecho se halló un gasto cardiaco de 14,2 L/min, con resistencia vascular sistémica de 574 dinas x s x cm-5 y presión sistólica de la arteria pulmonar en 46 mm Hg con media de 31 mm Hg. La ecografía abdominal mostró riñones de tamaño normal con adecuada diferenciación corticomedular; hígado aumentado de tamaño, con contornos irregulares y ecogenicidad heterogénea, sugestivos de cirrosis.
Se concluyó que el paciente presentó una falla cardiaca de alto débito por hiperflujo ocasionado por las múltiples fístulas arteriovenosas sistémicas, con gran dilatación de las cavidades derechas por sobrecarga crónica de volumen. Se inició tratamiento con diuréticos de asa, pero presentó mala diuresis con deterioro de la función renal y requirió inicio de terapia de reemplazo renal con hemodiálisis. Durante su hospitalización presentó varios episodios de epistaxis severa y requirió taponamiento nasal anterior y posterior por Otorrinolaringología.
Se solicitó evaluación por Radiología intervencionista para oclusión de fístulas arteriovenosas de pulmón y luego evaluación a Hepatología para manejo de la cirrosis y considerar posible trasplante hepático. No obstante, no pudieron realizarse dichos procedimientos porque el paciente presentó fibrilación auricular con contraindicación evidente para anticoagulación por sangrado activo anemizante y alto riesgo de sangrado por comorbilidades; posteriormente tuvo varios episodios de isquemia cerebral transitoria. Recibió tratamiento con ácido acetilsalicílico, pero empeoró la epistaxis por lo que debió suspenderse. Finalmente sufrió deterioro neurológico severo y se documentó enfermedad cerebrovascular isquémica que lo llevó a la muerte.
Discusión
La falla cardiaca de alto débito se define por la presencia de síntomas clásicos del síndrome asociado a gasto cardiaco por encima de lo normal, usualmente mayor de 8 L/min o un índice cardiaco mayor de 3,5 L/min/m2. Diversas condiciones nosológicas pueden llevar a la manifestación clínica en presencia de un corazón estructuralmente normal, básicamente por dos mecanismos: vasodilatación o aparición de fístulas arteriovenosas, que llevan a disminución de la resistencia vascular sistémica, activación neurohormonal y aumento del gasto cardiaco para sostener la perfusión periférica. Como causantes pueden mencionarse la anemia crónica, la hipercapnia crónica, la tirotoxicosis, la sepsis, el beriberi, el embarazo, la obesidad, la enfermedad hepática y el síndrome carcinoide por el mecanismo de vasodilatación, y la enfermedad de Paget, la enfermedad de Albrights, la enfermedad hepática y el síndrome carcinoide por la formación de fístulas arteriovenosas. Otras causas se descartan rápidamente con análisis sencillos como anemia, en el caso que se expone asociada a ferropenia por pérdida crónica de sangre, e hipertiroidismo, deficiencia de tiamina o policitemia, las cuales fueron excluidas en el paciente. También deben tenerse en cuenta otras causas de fístulas arteriovenosas viscerales como el antecedente de biopsias de órgano sólido, el trauma y los cateterismos. El 15% de los pacientes con THH no presentan historia de epistaxis ni lesiones cutáneas (2). Por esto ante un paciente con falla cardiaca de alto débito en quien no se encuentre ninguna de las causas descritas anteriormente, deben buscarse fístulas arteriovenosas viscerales asociadas a THH. Aunque es poco común que sea la primera manifestación de la THH, existen casos descritos en la literatura (3).
En la THH, la falla cardiaca de alto débito siempre se relaciona con fístulas arteriovenosas viscerales hepáticas y la edad promedio de presentación es de 59 años (2). Las fístulas arteriovenosas viscerales encontradas en hígado y pulmón generaron un cortocircuito de izquierda a derecha que produjo sobrecarga de volumen sobre las cavidades derechas, las cuales se dilataron de forma lenta y progresiva, produciendo clínicamente una falla derecha crónica con ingurgitación yugular, hepatomegalia, ascitis y edemas. El lado izquierdo del corazón se encontró hiperdinámico para expulsar dos o más veces su gasto cardiaco habitual, 14,2 L/min, con función sistólica preservada y la capacidad regenerativa intacta, como lo demostró la ecocardiografía transtorácica. Los parámetros hemodinámicos mostraron aumento del gasto cardiaco, con disminución de las resistencias vasculares e hipertensión pulmonar leve, hallazgos característicos de la enfermedad (4-6). El tratamiento médico se limitó al uso de diuréticos de asa para el manejo de los edemas y hemodiafiltración con buena respuesta. Los vasodilatadores como los inhibidores de la enzima convertidora de angiotensina, los antagonistas de los receptores de angiotensina II y los betabloqueadores están contraindicados y pueden producir hipotensión severa por las resistencias vasculares sistémicas que ya se encuentran muy bajas, por lo que no fueron utilizados. Los inotrópicos también empeoran la situación debido al estado hiperdinámico que tiene el ventrículo izquierdo (4).
En el año 2000 se publicaron los criterios del Consenso de Curaçao, para el diagnóstico clínico de THH (7). Según esta clasificación, un paciente tendrá diagnóstico "definitivo" si presenta 3 criterios, "posible" si cumple 2 criterios e "improbable" si presenta 0-1 criterios (Tabla 1). Este caso se clasificó como definitivo dada la presencia de los tres primeros criterios, sin una historia familiar clara. El examen genético busca mutaciones en los genes ENG y ACVRL1, aunque en este caso no fueron investigadas. En caso de ser negativo, se puede buscar mutación en SMAD4. En algunos casos no se encuentra ninguna alteración genética (2).

Las fístulas arteriovenosas viscerales hepáticas comprometen entre el 32% y el 78% de los pacientes con THH. Los síntomas se presentan en el 8% de los casos (8). Las formas de aparición más frecuentes son: falla cardiaca de alto débito, hipertensión portal y necrosis de vías biliares. También se puede presentar encefalopatía portosistémica y síndrome de robo intestinal. El diagnóstico pudo establecerse con ultrasonido Doppler y tomografía axial computarizada trifásica, aunque puede ser útil en algunos casos la imagen de resonancia magnética y la angiografía mesentérica. En la angiografía pueden observarse telangiectasias, malformaciones vasculares confluentes, dilatación de la arteria hepática y cortocircuitos arterioportales, arteriovenosos y/o portovenosos (9). En estudios no controlados se ha tratado el compromiso hepático con embolización de la arteria hepática y con trasplante hepático. La embolización produce mejoría significativa de los síntomas de falla cardiaca, pero su efecto generalmente es transitorio. El 30% de los pacientes embolizados presenta complicaciones graves como colangitis isquémica, colecistitis isquémica y/o necrosis hepática que llevan a muerte o trasplante hepático (1), por lo que esta opción terapéutica no fue contemplada. El trasplante hepático como tratamiento primario tiene una supervivencia a cinco años del 83% a pesar de un postoperatorio más complicado que el de trasplantes por otras causas por mayor sangrado, transfusiones y estancia hospitalaria (10). Las guías de la Fundación Internacional para THH recomiendan remitir los pacientes para trasplante hepático en tres condiciones: necrosis biliar isquémica, falla cardiaca intratable e hipertensión portal intratable (1). En este caso en particular fue contemplada la posibilidad, sin embargo dadas las condiciones clínicas y la no resolución de las fístulas arteriovenosas viscerales pulmonares, se decidió posponer la decisión de la intervención.
La epistaxis es la manifestación más frecuente de la enfermedad. Puede producir sangrados severos y difíciles de controlar que llegan a causar anemia y requerir soporte transfusional. El tratamiento de elección es la cauterización endonasal, que debe ser hecha por expertos ya que es frecuente que se presenten perforaciones septales (1). En casos refractarios pueden hacerse procedimientos invasivos como la dermoplastia septal o procedimiento de Young (11). Cuando se requiere taponamiento nasal, este debe hacerse con material atraumático e idealmente neumático, el cual se desinfla al retiro para evitar mayor lesión.
Cerca del 1% de los pacientes con THH desarrollan hipertensión pulmonar no relacionada con fístulas arteriovenosas viscerales. El comportamiento puede ser similar al de la hipertensión pulmonar idiopática y se asocia a mutaciones en el gen ACVRL1 que codifica ALK1(12). Más frecuentemente, la hipertensión pulmonar está relacionada con fístulas arteriovenosas viscerales hepáticas y falla cardiaca de alto débito. Las fístulas arteriovenosas viscerales en el pulmón se presentan entre el 15% y el 50% de los pacientes. Menos del 10% muestra signos clínicos relacionados con el cortocircuito derecha-izquierda como soplos, cianosis e hipocratismo digital. Su presencia se acompaña de complicaciones como enfermedad cerebrovascular embólica, abscesos cerebrales, hemoptisis masiva y hemotórax espontáneo (13). La ecocardiografía transtorácica con solución salina agitada es el método de tamizaje de elección para detectar fístulas arteriovenosas viscerales en el pulmón, con una sensibilidad del 97% y valor predictivo negativo del 99% (14-17). Para una adecuada definición anatómica y definir una posible oclusión percutánea se emplean la angiotomografía axial computarizada y la arteriografía. La embolización selectiva de las fístulas está indicada en fístulas arteriovenosas viscerales sintomáticas mayores de 2 centímetros o en asintomáticas mayores de 3 centímetros, logrando un éxito hasta en el 93% de los casos de forma inmediata. Puede haber revascularización hasta en el 15% a 10 años, y 18% de los pacientes desarrolla microfístulas; no obstante, las complicaciones graves son raras (18). Se consideró la posibilidad de realizar cierre con tapón vascular tipo Amplatzer; sin embargo, el procedimiento no se llevó a cabo inicialmente por la falla renal y posteriormente por la enfermedad cerebrovascular y el deterioro clínico. El seguimiento se realiza con angiotomografía ya que la ecocardiografía con contraste salino persiste positiva de forma indefinida a pesar del cierre (15). Se recomienda que estos pacientes reciban profilaxis con antibióticos previo a procedimientos de alto riesgo de bacteriemia. También se recomienda abstenerse de la práctica del buceo (1).
Otros órganos comprometidos son el tracto gastrointestinal que puede presentar telangiectasias hasta en el 80% de los casos, pero los sangrados clínicos sólo ocurren en el 25% de los pacientes durante la quinta o sexta décadas de la vida. Pueden desarrollarse malformaciones arteriovenosas cerebrales hasta en el 23% de los pacientes con HHT; adicionalmente, el riesgo de sangrado anual se estima en 0,5%.
Conclusiones
La THH es una entidad poco frecuente y de manifestaciones tan variables que hacen que su diagnóstico en ausencia de epistaxis y telangiectasias en piel y mucosas, sea difícil. En casos de falla cardiaca de alto débito con dilatación severa de las cavidades derechas, siempre debe tenerse presente la posibilidad de fístulas arteriovenosas viscerales y THH. El caso descrito tuvo diagnóstico definitivo según los criterios de Curaçao a pesar de no tener antecedentes familiares. La mayoría de las complicaciones en la THH, se presentan después de la cuarta década de la vida debido a que las telangiectasias y las MAV aumentan en tamaño y cantidad con el paso de los años. El tratamiento médico no modifica la historia natural de la falla cardiaca de alto débito y siempre debe corregirse la causa de base. En este paciente debía considerarse el cierre de las fístulas arteriovenosas viscerales del pulmón por radiología intervencionista, a pesar del riesgo de aumentar la hipertensión pulmonar por el cortocircuito de izquierda a derecha producido por las fístulas arteriovenosas viscerales en el hígado. Este procedimiento está contraindicado en el hígado por la alta probabilidad de complicaciones graves y los pacientes con falla cardiaca intratable deben someterse a trasplante hepático, luego del cual se ha descrito regresión completa de la falla cardiaca. Los eventos cerebrovasculares son una complicación descrita en pacientes con fístulas arteriovenosas viscerales del pulmón, posiblemente por embolismo paradójico; sin embargo en este paciente estuvieron asociados a la presencia de fibrilación auricular, que fue lo que tal vez provocó su muerte.
Bibliografía
1. Faughnan ME, Palda VA, Garcia-Tsao G, Geisthoff UW, McDonald J, Proctor DD, et al. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J Med Genet. 2011; 48 (2): 73-87. [ Links ]
2. Trembath RC, Thomson JR, Machado RD, Morgan NV, Atkinson C, Winship I, et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med. 2001; 345 (5): 325-334. [ Links ]
3. Montejo Baranda M, Perez M, De Andres J, De la Hoz C, Merino J, Aguirre C. High out-put congestive heart failure as first manifestation of Osler-Weber-Rendu disease. Angiology. 1984; 35 (9): 568-576. [ Links ]
4. Mehta PA, Dubrey SW. High output heart failure. QJM. 2009; 102 (4): 235-241. [ Links ]
5. Clayton T, Banks KP, Bui-Mansfield LT. AJR teaching file: High-output cardiac failure in a patient with a history of hereditary hemorrhagic telangiectasia. AJR Am J Roentgenol. 2006; 187 (6 Suppl.): S508-510. [ Links ]
6. Brohée D, Franken P, Fievez M, Baudoux M, Hénuzet C, Brasseur P, et al. High-output right ventricular failure secondary to hepatic arteriovenous microfistulae. Selective arterial embolization treatment. Arch Intern Med. 1984; 144 (6): 1282-1284. [ Links ]
7. Shovlin CL, Guttmacher AE, Buscarini E, Faughnan ME, Hyland RH, Westermann CJ, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet. 2000; 91(1): 66-67. [ Links ]
8. Buscarini E, Plauchu H, Garcia Tsao G, White RI Jr, Sabbà C, Miller F, et al. Liver involvement in hereditary hemorrhagic telangiectasia: consensus recommendations. Liver Int. 2006; 26 (9): 1040-6. [ Links ]
9. Garcia-Tsao G, Korzenik JR, Young L, Henderson KJ, Jain D, Byrd B, et al. Liver disease in patients with hereditary hemorrhagic telangiectasia. N Engl J Med. 2000; 343 (13): 931-6. [ Links ]
10. Lerut J, Orlando G, Adam R, Sabbà C, Pfitzmann R, Klempnauer J, et al. Liver transplantation for hereditary hemorrhagic telangiectasia: Report of the European liver transplant registry. Ann Surg. 2006; 244 (6): 854-62. [ Links ]
11. Nasal cavity dermoplasty as a treatment of recurr... [Otolaryngol Pol. 2007] - PubMed result [Internet]. [cited 2011 Abr. 30]. Disponible en:: http://www.ncbi.nlm.nih.gov/pubmed/17605423. [ Links ]
12. Cottin V, Dupuis-Girod S, Lesca G, Cordier J-F. Pulmonary vascular manifestations of hereditary hemorrhagic telangiectasia (Rendu-Osler disease). Respiration. 2007;74(4):361-78. [ Links ]
13. Faughnan ME, Granton JT, Young LH. The pulmonary vascular complications of hereditary haemorrhagic telangiectasia. Eur Respir J. 2009; 33 (5): 1186-94. [ Links ]
14. Barzilai B, Waggoner AD, Spessert C, Picus D, Goodenberger D. Two-dimensional contrast echocardiography in the detection and follow-up of congenital pulmonary arteriovenous malformations. Am J Cardiol. 1991; 68 (15): 1507-10. [ Links ]
15. Lee WL, Graham AF, Pugash RA, Hutchison SJ, Grande P, Hyland RH, et al. Contrast echocardiography remains positive after treatment of pulmonary arteriovenous malformations. Chest. 2003; 123 (2): 351-8. [ Links ]
16. Van Gent MWF, Post MC, Luermans JGLM, Snijder RJ, Westermann CJJ, Plokker HWM, et al. Screening for pulmonary arteriovenous malformations using transthoracic contrast echocardiography: a prospective study. Eur Respir J. 2009; 33 (1): 85-91. [ Links ]
17. Nanthakumar K, Graham AT, Robinson TI, Grande P, Pugash RA, Clarke JA, et al. Contrast echocardiography for detection of pulmonary arteriovenous malformations. Am Heart J. 2001; 141 (2): 243-6. [ Links ]
18. Haitjema T, ten Berg JM, Overtoom TT, Ernst JM, Westermann CJ. Unusual complications after embolization of a pulmonary arteriovenous malformation. Chest. 1996; 109 (5): 1401-4. [ Links ]
