










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Cardiología
Print version ISSN 0120-5633
Rev. Colomb. Cardiol. vol.21 no.1 Bogota Jan./Feb. 2014
(1) Grupo de Investigación en Genética Humana Aplicada. Facultad de Ciencias de la Salud, Universidad del Cauca, Popayán, Colombia.
(2) Unidad Vascular, Popayán, Colombia.
(3) Fundación InnovaGen, Popayán, Colombia.
Correspondencia: Dr. Carlos H. Sierra, Laboratorio de Genética Humana, Facultad de Ciencias de la Salud, Universidad del Cauca, Calle 5 No. 4-70, Popayán, Colombia. Telefax: (572) 8 20 98 72. Correo electrónico: hsierra@unicauca.edu.co
Recibido: 24/05/2013. Aceptado: 22/08/2013.
La aterosclerosis es el resultado de la alteración en la función del endotelio arterial, desencadenada por la exposición continua de este tejido a fenómenos circulatorios turbulentos. La presencia de factores de riesgo cardiovascular promueve la sobre-expresión de moléculas proinflamatorias que inician la cascada inflamatoria al interior del vaso. Una vez las células inmunes, como monocitos y macrófagos, ingresan a la arteria, se inicia una serie de eventos que incluye la internalización de partículas lipídicas en el macrófago y la formación de las células espumosas y estrías grasas. Posteriormente, la respuesta inflamatoria se agudiza y continúa la formación del núcleo lipídico y el desarrollo de la placa de ateroma. El proceso inflamatorio modula la sobre-expresión de mecanismos protrombóticos que actúan en respuesta a la ruptura o erosión de la placa aterosclerótica y desencadena eventos trombóticos o embólicos. El objetivo de esta revisión es presentar evidencia acerca de los mecanismos celulares y moleculares involucrados en los procesos de disfunción endotelial, inflamación y trombosis que subyacen el inicio y la progresión de la aterosclerosis.
PALABRAS CLAVE: radicales libres, endotelio, inflamación, coagulación, aterosclerosis, enfermedad cardiovascular
Atherosclerosis results from an altered arterial endothelial function, triggered by the continuous exposure of this tissue to turbulent circulatory phenomena. The presence of cardiovascular risk factors promotes the overexpression of proinflammatory molecules that initiate the inflammatory cascade within the vessel. Once immune cells such as monocytes and macrophages have entered the artery, these initiate a series of events that include the internalization of lipid particles in the macrophage and the formation of foam cells and fatty streaks. Subsequently, the inflammatory response is exacerbated and the lipid core formation and development of atheromatous plaque continues. The inflammatory process modulates the overexpression of prothrombotic mechanisms that act in response to the rupture or erosion of the atherosclerotic plaque and triggers thrombotic or embolic events. The aim of this review is to present evidence about the cellular and molecular mechanisms involved in the processes of endothelial dysfunction, inflammation and thrombosis that underlie the initiation and progression of atherosclerosis.
KEYWORDS: oxidative stress, endothelial dysfunction, inflammation, coagulation, atherosclerosis, cardiovascular disease.
Introducción
Actualmente, las enfermedades cardiovasculares constituyen la principal causa de muerte y discapacidad a largo plazo (1). Según reportes epidemiológicos del año 2008, alrededor de 7 millones de personas murieron a causa de infarto agudo del miocardio y 6,15 millones a causa de accidente cerebrovascular, constituyendo el 23,6% de mortalidad mundial atribuible a este tipo de enfermedades (2). En Colombia las enfermedades cardiovasculares constituyen la principal causa de muerte no violenta; de éstas la enfermedad isquémica es responsable de 83,7 defunciones por cada 100.000 habitantes y la enfermedad cerebrovascular de 42,6 muertes/100.000 habitantes (3).
La aterosclerosis es la principal causa de enfermedades cardiovasculares, caracterizada principalmente por la acumulación de células inflamatorias, lipoproteínas y tejido fibroso en la túnica íntima arterial (4). La enfermedad inicia en la niñez y progresa en un estado subclínico, y eventualmente se manifiesta pasada la cuarta década de la vida (5). El engrosamiento progresivo de la pared arterial debido a la formación de placas ateroscleróticas, conlleva complicaciones cardiovasculares a causa de la formación de trombos en las placas, y ocasiona así la estenosis de la luz vascular y la consecuente necrosis del tejido afectado (6, 7).
Los procesos fisiopatológicos que conducen a la aterosclerosis son desencadenados por factores de riesgo cardiovasculares modificables, tales como la diabetes mellitus, el tabaquismo, la hipertensión arterial, la dislipidemia y la hiperhomocisteinemia, y no modificables como la edad, el género, la historia familiar y la susceptibilidad genética (8). Adicionalmente, la identificación de características como los niveles de lipoproteína anormal y fibrinólisis, ejerce un papel importante en el inicio y la progresión de la aterosclerosis (9). Los factores de riesgo median los mecanismos moleculares y celulares asociados a estrés oxidativo, inflamación y coagulación, principales vías patológicas precursoras de la enfermedad aterosclerótica (10). Además, la interacción de factores intrínsecos (i.e. susceptibilidad del individuo) y extrínsecos (i.e. medio ambiente) determinan la aparición de la enfermedad y posibilitan la identificación de fenotipos asociados a la progresión de la enfermedad (11, 12).
A pesar del problema de salud pública que representa la aterosclerosis dada la carga de enfermedad, el conocimiento existente acerca de las causas o factores involucrados en su desarrollo es poco concluyente. Sin embargo, durante la última década se ha podido demostrar que los eventos tempranos de la enfermedad están asociados con una disfunción en el endotelio vascular, seguida por la formación de la estría grasa y la aparición posterior de procesos aterotrombóticos (13). El objetivo de esta revisión es presentar evidencia actualizada de los principales mecanismos celulares y moleculares que ejercen un rol crítico en el inicio y establecimiento de fenómenos aterotrombóticos. Se plantean como vías patológicas precursoras de la aterosclerosis el estrés oxidativo asociado a disfunción endotelial, la inflamación asociada a la formación y progresión de la placa de ateroma y la coagulación relacionada con la aparición de eventos trombóticos cardiovasculares.
Estrés oxidativo y disfunción endotelial
El endotelio arterial libera diversas sustancias que mantienen la función arterial controlando mecanismos de relajación y contracción vascular, trombogénesis y fibrinólisis, y la activación e inhibición de mecanismos inflamatorios (14, 15). El balance hemostático mantiene el tejido endotelial en un estado quiescente caracterizado por adhesión leucocitaria baja, niveles de radicales superóxido bajos, biodisponibilidad de vasodilatadores significativa y permeabilidad baja (16). Cuando el control del tejido endotelial se ve afectado por la presencia de flujos turbulentos y concentraciones elevadas de especies reactivas de oxígeno (EROs), su función se altera permitiendo la sobre-producción de radicales superóxido, la baja disponibilidad de óxido nítrico (ON) y la expresión de mecanismos pro-apoptóticos y pro-trombóticos (17, 18).
El estrés oxidativo se reconoce como un regulador clave de procesos fisiológicos y fisiopatológicos de las células vasculares como la oxidación lipídica y de proteínas en la pared del vaso arterial (19, 20) y por ende, es considerado un evento temprano en la aterogénesis. El estrés oxidativo promueve la reducción de ON y la sobre-expresión de moléculas inflamatorias (21). Además, la producción de radicales H2O2 media la proliferación de células del músculo liso vascular inducido por el factor de crecimiento derivado de plaquetas (22). Las EROs son igualmente importantes en la activación de las plaquetas en la respuesta trombótica temprana a la injuria vascular, representando mecanismos claves en el inicio de fenómenos aterotrombóticos (23).
Las EROs tienen múltiples efectos en la señalización, la modificación de moléculas y los daños a los sistemas biológicos, basados en su capacidad de interactuar con componentes celulares específicos (24). Así, la función del estrés oxidativo en los diferentes procesos fisiopatológicos de la enfermedad aterosclerótica, lo convierten en un blanco importante para la implementación de estrategias de prevención (e.g. manejo de factores de riesgo) y el desarrollo de nuevos fármacos.
Óxido nítrico y disfunción endotelial
El ON es el vasodilatador más importante producido por el endotelio arterial. Se forma a partir de la L-arginina vía acción enzimática de la óxido nítrico sintasa (eNOS) y cofactores como la tetrahidrobiopterina y la nicotinamida adenina dinucleótido fosfato (NADPH) que facilitan su síntesis (25, 26) (figura 1). La disfunción endotelial se origina a partir de la pérdida del ON y la sobreproducción de vasoconstrictores, que desencadenan la regulación afectada de mecanismos proinflamatorios e incrementan la permeabilidad arterial, el crecimiento celular en la pared arterial y el estado hipercoagulante (27).
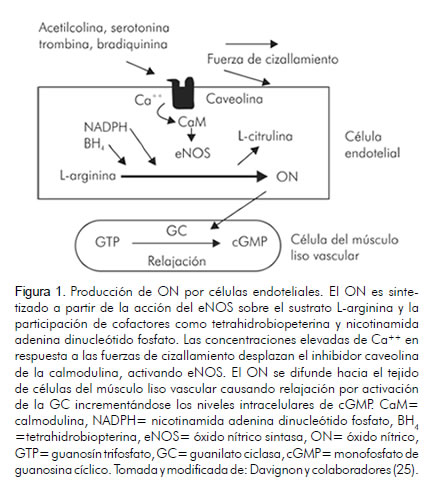
La alteración en la funcionalidad endotelial es el resultado de la apoptosis y el estado protrombótico de la célula endotelial, eventos originados a partir de la exposición constante del tejido arterial a elevadas concentraciones de EROs (28, 29). La sobreproducción de EROs es promovida por la presencia de factores de riesgo cardiovascular, principalmente tabaquismo, hiperhomocisteinemia, dislipidemia y diabetes mellitus, que contribuyen al daño continuo en la pared arterial (30) y establecen un estado constante de estrés oxidativo (figura 2).
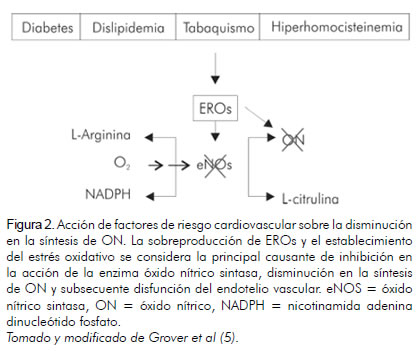
El tabaquismo promueve la producción de EROs debido a que compuestos como los hidrocarbonos aromáticos policíclicos y la nicotina, reducen la producción y disponibilidad del ON (31). En pacientes con hiperhomocisteinemia, la concentración elevada de homocisteína afecta la biodisponibilidad del ON debido al agotamiento del vasodilatador ya constituido, a causa de su reacción con homocisteína para formar S-nitrosohomocisteína (32). Por otro lado, los niveles elevados de colesterol y triglicéridos, observados en pacientes dislipidémicos, median la sobre-regulación de la enzima NADPH oxidasa y la producción de radicales O2- (33).
En personas diabéticas se ha observado la sobre-regulación de la inflamación y la sobreproducción de EROS que incrementan la peroxidación lipídica, inhibiendo la acción de la eNOS y la producción de ON y reduciendo la respuesta en tejidos blanco (27). La hiperglicemia promueve la sobreproducción de EROs, resultado de la glicación de lípidos y proteínas intra y extracelulares que lideran la generación de productos finales de glicación (AGEs, su sigla en inglés) (24). Los AGEs se unen a sus receptores RAGEs situados en macrófagos, monocitos y células del músculo liso vascular, amplificando la respuesta inflamatoria, el incremento de la permeabilidad vascular y el estrés oxidativo (34). La disminución en la producción de ON observada en pacientes diabéticos obedece a la activación de la proteína quinasa C, cuya expresión reduce la actividad enzimática de eNOS y el incremento en la producción de vasoconstrictores como la endotelina-1 (35). Igualmente, la proteína quinasa C mejora la expresión de moléculas de adhesión a la pared del vaso, mediando así procesos de activación del endotelio arterial (36).
Activación del endotelio e inicio de la formación de la placa
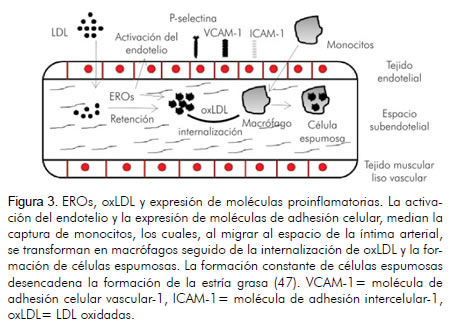
La activación del endotelio involucra la expresión de moléculas de adhesión celular como la molécula de adhesión intercelular-1 (ICAM-1) y la molécula de adhesión vascular endotelial-1 (VCAM-1) (37), facilitando el reclutamiento de células inflamatorias y la deposición constante de lípidos en la capa íntima de la arteria. Además, la permeabilidad de la pared arterial media la retención de partículas de lipoproteínas de baja densidad (LDL) químicamente modificadas (38). Las modificaciones químicas de las LDL ocurridas en el espacio subendotelial arterial, corresponden principalmente a procesos de acetilación y oxidación (39). La oxidación de las LDL se asocia con procesos aterogénicos que median el reclutamiento de monocitos a la íntima arterial; mejoran la tasa de captura de lipoproteínas y favorecen la continuidad del proceso inflamatorio y la expresión de mecanismos protrombóticos (figura 3) (40, 41). Adicionalmente, las oxLDL, la constante expresión de moléculas de adhesión y la retención y oxidación de partículas de LDL, promueven la expresión de citoquinas, quimiocinas y selectinas, mediando procesos inflamatorios en la pared arterial (42, 43), agudizando así la respuesta inmune y conformando el inicio de estrías grasas, que constituyen la primera lesión visible de la enfermedad aterosclerótica.
Mecanismos inflamatorios y formación de la placa
Cascada de adhesión leucocitaria y transmigración al espacio subendotelial
La cascada de adhesión leucocitaria tradicional involucra la captura, el rodamiento, la activación, la adhesión y la migración transendotelial (44). Cada uno de estos pasos en la cascada de adhesión está mediado por moléculas de adhesión, las cuales están divididas en cuatro grandes familias: 1) selectinas, 2) ligandos de selectinas, 3) inmunoglobulinas (ICAM-1 y VCAM-1), y 4) integrinas (45). La unión de selectinas (expresadas en la pared del vaso) y ligandos de selectinas (presentes en las células inmunes) median la atracción y el rodamiento de las células, mientras que las moléculas pertenecientes a la familia de las inmunoglobulinas y las integrinas están involucradas en la adhesión y migración leucocitaria al interior del vaso (46, 47).
La migración de los leucocitos se genera en respuesta a la acción de quimiocinas como la proteína quimioatrayente de monocitos (MCP-1) y los ligandos 5 y 2 de quimiocina y fractalquina (39). La diapédesis de leucocitos al interior de la pared vascular requiere un gradiente quimiotáctico ejercido por concentración de quimiocinas; igualmente, la expresión de las mismas modula las características adhesivas de las integrinas y apoya el reclutamiento de leucocitos (48).
El ingreso de monocitos al espacio subendotelial de la arteria, alienta la producción del factor estimulante de colonias de macrófagos (M-CSF), el cual promueve la conversión de monocitos en macrófagos (49). La diferenciación de los monocitos puede darse igualmente en células dendríticas, pero depende de las condiciones microambientales, así como del contenido y la composición de las citoquinas presentes en la placa en formación (50).
Algunos subconjuntos de monocitos (principalmente CD14+CD16-CCR2+ y CD14-CD16+CX3CR1+) que ingresan en la íntima arterial en estados inflamatorios agudos, se observan dentro de la placa aterosclerótica y presentan diversos marcadores y comportamientos durante la inflamación (51, 52). Recientemente se ha descubierto que estas células tienen papeles complementarios durante la progresión de la AR (53). Hasta el momento, altas concentraciones del subconjunto monocitario CD14+CD16-CCR2+ se han asociado de manera significativa con un espesor incrementado de la íntima-medial carotídea y con una alta vulnerabilidad de placas ateroscleróticas coronarias (54).
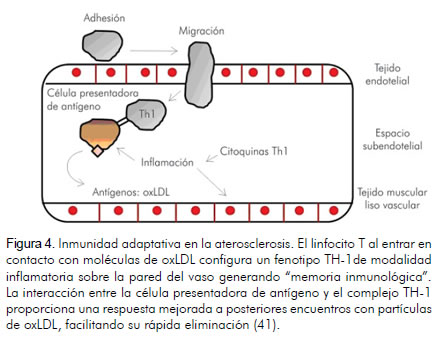
Otras células inflamatorias, como las células dendríticas y los linfocitos T al interior de la placa, se distribuyen mayormente en áreas de neovascularización, la cual representa el crecimiento de la vasa vasorum dentro de la arteria, característico de placas susceptibles de ruptura (50, 55). Adicionalmente, la continua interacción de linfocitos T con células presentadoras de antígenos y la participación en procesos de inmunidad adaptativa (figura 4), abre nuevos horizontes para la identificación y validación de biomarcadores propios de este proceso (56), fenómeno que permitiría la estratificación del riesgo en las poblaciones e incluso el desarrollo de vacunas para blancos moleculares que facilitarían la prevención primaria de eventos cardiovasculares (57).
Formación de células espumosas y núcleo lipídico necrótico
Los macrófagos expresan en su superficie celular una serie de receptores específicos para oxLDL denominados receptores "basurero"; incluyendo los receptores CD36, CD68 y el receptor 1 de lipoproteína de baja densidad oxidada (58). A causa que los fosfolípidos oxidados, principales componentes estructurales de las oxLDL, que son ligandos de estos receptores, el colesterol es internalizado hacia los macrófagos, y las enzimas lipolíticas degradan el colesterol a ésteres de colesterol (59). Estos últimos son hidrolizados al interior citoplasmático de los macrófagos para liberar el colesterol no esterificado y se transfieren al colesterol de la lipoproteína de alta densidad por medio del casete transportador unido a adenosín trifosfato y a la proteína de transferencia de colesterol, mecanismo conocido como transporte reverso del colesterol (60).
La continua internalización de oxLDL desencadena la formación de cuerpos apoptóticos y la interacción de AGEs con células inflamatorias- en el caso del paciente diabético agudizan la respuesta inflamatoria con la sobreexpresión del factor nuclear kappa-B (NF-kB) (61), el cual regula la producción y expresión de citoquinas proinflamatorias, principalmente factor de necrosis tumoral (TNF-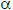 ) (62), interferón γ e interleuquinas (IL) 1, 6 y 18, que alteran la distribución de complejos caderina-catenina del endotelio vascular, al tiempo que reestructuran las uniones intercelulares, permeabilizan el tejido endotelial y facilitan el ingreso de mayor cantidad de partículas de LDL y la adhesión-transmigración de leucocitos (63). La destrucción de las células espumosas está acompañada por la acumulación de lípidos y de oxLDL que pueden causar daño extenso en el ADN y perpetuar la muerte celular (64).
) (62), interferón γ e interleuquinas (IL) 1, 6 y 18, que alteran la distribución de complejos caderina-catenina del endotelio vascular, al tiempo que reestructuran las uniones intercelulares, permeabilizan el tejido endotelial y facilitan el ingreso de mayor cantidad de partículas de LDL y la adhesión-transmigración de leucocitos (63). La destrucción de las células espumosas está acompañada por la acumulación de lípidos y de oxLDL que pueden causar daño extenso en el ADN y perpetuar la muerte celular (64).
La acción del NF-kB también influye en la regulación genética del ciclo de las células del músculo liso vascular, promoviendo un fenotipo proliferativo y migratorio de la capa media de la arteria hacia la capa íntima, rodeando el núcleo lipídico necrótico y conformando el fibroateroma (65). Adicionalmente, la acción del NF-kB promueve la expresión de metaloproteinasa-1, potente colagenasa implicada en la degradación de colágeno, componente de la matriz extracelular, y le confiere una alta susceptibilidad de ruptura a una placa aterosclerótica inestable (66).
Mecanismos de coagulación y trombosis arterial
La morfología de la placa es clave en el desarrollo de la trombosis arterial y en la aparición de enfermedades cardiovasculares (67). Las lesiones ateroscleróticas típicas están compuestas por un núcleo lipídico rodeado de una capa fibrosa compuesta principalmente por células del músculo liso vascular y colágeno que le da resistencia e impide la ruptura de la placa y los posibles eventos trombóticos subsecuentes (68).
Se ha descrito que la vulnerabilidad de la placa a la ruptura depende de características como el tamano y la consistencia del núcleo ateromatoso, los niveles de colágeno reducidos y la capa fibrosa delgada, así como la inflamación al interior de la placa y la "fatiga" de la cubierta fibrosa (69). Las placas estables se caracterizan por presentar mayor disfunción y permeabilidad endotelial, mayor activación plaquetaria y reclutamiento leucocitario y mejor remodelado vascular. Las placas inestables presentan más procesos relacionados con angiogénesis, mayor estrés oxidativo y apoptosis, así como mayores tasas de proteólisis e inflamación (70).
La trombosis arterial constituye un proceso patológico dinámico, resultado de una alteración en las funciones del endotelio arterial, el cual adopta un fenotipo vasoconstrictor, procoagulante, activador plaquetario e inhibidor de procesos anti-fibrinolíticos (71). Los procesos inflamatorios subsecuentes a la activación del endotelio incrementan la expresión de moléculas protrombóticas como el factor tisular, inhiben la actividad fibrinolítica y alteran las propiedades antiagregantes y vasodilatadoras del endotelio (72). La interacción molecular de vías patológicas de trombosis y fibrinólisis, media el inicio y la progresión de un evento agudo trombótico dentro de lesiones aterotrombóticas
Hemostasia vascular y mecanismos protrombóticos
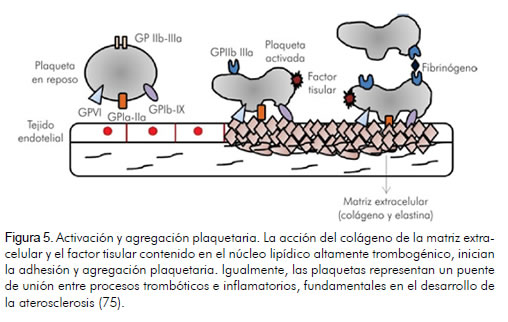
El sistema hemostático vascular es ejecutado a través de una red de procesos que incluyen sistema plaquetario, coagulación y vías anticoagulantes y fibrinolíticas (70). Las plaquetas interactúan con el tejido endotelial vascular y están ligadas a mecanismos inflamatorios, trombosis y enfermedades cardiovasculares a través de interacciones moleculares entre receptores del tejido endotelial, plaquetas y leucocitos (73). Luego de la ruptura y el daño en la pared arterial, proteínas de la matriz extracelular altamente reactivas a plaquetas, incluyendo colágeno, factor Von Willebrand (vWF), fibronectina y laminina, al ser expuestas al torrente sanguíneo, mejoran la expresión de receptores en la superficie plaquetaria (74, 75) (figura 5).
La producción de factor tisular es dependiente de IL-6, angiotensina II, oxLDL y TNF-, y constituye un paso importante en la generación de trombina (76), que actúa suprimiendo dos péptidos de bajo peso molecular del fibrinógeno, dando lugar a monómeros de fibrina que se aglomeran conformando y estabilizando el retículo del trombo (64, 77). Adicionalmente y subsecuente a la inducción del factor tisular, las citoquinas proinflamatorias (principalmente IL-1 y TNF-) y los neutrófilos activados afectan las vías anticoagulantes naturales (antitrombina, sistema de proteína C e inhibidor de la vía del factor tisular) (78).
La trombina es uno de los estimuladores más potentes de activación plaquetaria a través de los receptores plaquetarios activados por proteasas, específicamente las clases 1 y 4 en plaquetas humanas (79), moléculas que también participan en la respuesta pro-inflamatoria observada en la AR y la restenosis. Además, la acción de la trombina incrementa la expresión de IL-8, MCP-1 y E-selectina en células endoteliales que al unirse con receptores plaquetarios activados por proteasas clase 2, conllevan sobrerregulación de la respuesta inflamatoria, mayor producción de EROs, así como expresión de otras moléculas de adhesión celular (80).
Los fenómenos de activación y agregación plaquetaria continúan con el proceso trombótico. Uno de los factores relacionados con la susceptibilidad de eventos es la disfunción plaquetaria relacionada a la hiperreactividad de las plaquetas, fenómeno observado en pacientes con diabetes mellitus tipo 2 y acción defectuosa de la insulina (81). En los diabéticos, la adhesión y la agregación plaquetaria ocurre más fácilmente y los niveles de fibrinógeno son más elevados en comparación con los no diabéticos, de ahí que la enfermedad se considere como un estado hipercoagulante (82).
Activación, adhesión y agregación plaquetaria
La activación de las plaquetas es seguida por procesos de atracción, rodamiento y adhesión en la pared arterial dañada, donde la interacción de la plaqueta con el colágeno de la matriz extracelular, es mediada por la glicoproteína VI e integrina 2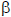 1 (83). La adhesión específica de las plaquetas es regulada por la interacción entre el receptor de la glicoproteína Ib/V/IX en la superficie plaquetaria y el vWF y entre la glicoproteína VI y el colágeno expresado en los sitios de injuria vascular (84).
1 (83). La adhesión específica de las plaquetas es regulada por la interacción entre el receptor de la glicoproteína Ib/V/IX en la superficie plaquetaria y el vWF y entre la glicoproteína VI y el colágeno expresado en los sitios de injuria vascular (84).
Una vez adheridas, las plaquetas liberan el contenido de los gránulos plaquetarios y sobre-regulan la función de ciertas moléculas adhesivas como la integrina IIb3, molécula que se une a múltiples ligandos entre ellos el vWF, el fibrinógeno, la fibrina y la fibronectina (85) indispensables para la formación estable de agregados plaquetarios; además, secretan otros mediadores aterogénicos como citoquinas, quimiocinas, factores de crecimiento, moléculas de coagulación y factores de la coagulación (70), mejorando la respuesta inflamatoria y protrombótica, característica de los fenómenos aterotrombóticos.
Conclusiones y perspectivas
La aterosclerosis se considera como una enfermedad multifactorial originada a partir de la interacción entre factores de riesgo que contribuyen a la aparición temprana de la misma, aumentando la probabilidad de padecer una enfermedad cardiovascular. Adicionalmente, la alteración de ciertos mecanismos moleculares y celulares promueve el desarrollo del proceso obstructivo arterial. Es evidente que el entendimiento de las interacciones entre los factores intrínsecos y extrínsecos y las rutas celulares y moleculares que contribuyen al desarrollo de estas patologías, generará nuevas avenidas de investigación para el conocimiento del origen, el inicio y la progresión de las enfermedades cardiovasculares. Se esperarían, entonces, nuevas estrategias de prevención avaladas en una mejor estratificación del riesgo en poblaciones afectadas a través del uso de biomarcadores (86), y a la vez, nuevos fármacos diseñados para controlar o modular blancos moleculares importantes involucrados en el proceso aterogénico (87).
Recientemente, se ha establecido la presencia de diversas proteínas del secretoma vascular que no son encontrados en el plasma y aún permanecen sin caracterizar. Estas sustancias específicamente se sintetizan al inicio de procesos de injuria y activación endotelial. La identificación de estas proteínas y la determinación de su expresión en arterias coronarias, pueden servir como herramienta para la realización de un posible diagnóstico temprano y posibilitarían la creación de estrategias entorno a la inhibición de mecanismos de daño endotelial, en poblaciones bajo riesgos significativos (88).
Las células endoteliales se encuentran expuestas continuamente a estímulos mecánicos y químicos que alteran su comportamiento y desencadenan su fenotipo disfuncional, cuya característica patológica más relevante corresponde a una disminución del ON. Las moléculas involucradas en la activación de las células endoteliales son críticas en el reclutamiento de linfocitos y se encuentran asociadas a inestabilidad y ruptura de la placa; además, son útiles como biomarcadores para la identificación de poblaciones en riesgo. La subsecuente inflamación en la pared arterial se halla promovida por inmunidad innata e inmunidad adaptativa, caracterizándose por la presencia de agregados celulares y citoquinas específicas que pueden ser encontrados en placas humanas o de modelos experimentales. Las estrategias basadas en vacunación serían capaces de promover la respuesta celular específica del antígeno, al usar como blanco la entrega de citoquinas anti-inflamatorias/anti-aterogénicas en la placa, y reducir potenciales efectos secundarios (56).
La placa de fibroateroma está compuesta por material lipídico, células inflamatorias y componentes de la matriz extracelular como colágeno y elastina. Aunque se ha abordado el papel de estas últimas en procesos de trombogénesis y características morfológicas de la placa, la información es escasa y poco concluyente. Sumado a ello, conocer ampliamente todos los mecanismos subyacentes alrededor de las múltiples funciones de estas moléculas y la formación de la parte fibrosa de la placa, brindará herramientas para facilitar la identificación de placas vulnerables y evitar complicaciones posteriores (89).
La expresión de metaloproteinasas en el endotelio vascular, está involucrada en remodelar o digerir cualquier componente extracelular del vaso. Sin embargo, las metaloproteinasas derivadas de las células endoteliales reciben menos atención que aquellas producidas por el tejido del músculo liso, constituyéndose en un blanco atractivo en futuras investigaciones, con la finalidad de esclarecer su función en procesos inflamatorios tempranos y tardíos de la enfermedad (90).
Agradecimientos
Los autores expresan su agradecimiento a la Universidad del Cauca, a la Vicerrectoría de Investigaciones y a los Departamentos de Ciencias Fisiológicas y Ciencias Quirúrgicas de la Facultad de Ciencias de la Salud por su apoyo en la ejecución de este proyecto.
Conflicto de interés: los autores declaran no tener conflicto de intereses.
Fuente de financiamiento: esta investigación fue financiada con recursos del Programa Nacional de Salud de Colciencias (Cod. 110351929119).
Bibliografía
1. Greenland P, Alpert JS, Beller GA, Benjamin EJ, Budoff MJ, Fayad ZA, et al. 2010 ACCF/AHA guideline for assessment of cardiovascular risk in asymptomatic adults. J Am Coll Cardiol. 2010; 56: 50-103. [ Links ]
2. Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, et al. Heart disease and stroke statistics-2013 update: a report from de American Heart Association. Circulation. 2013; 127: 6-245. [ Links ]
3. Lopez P, López J. Lecciones aprendidas de dos grandes estudios epidemiológicos de enfermedades cardio-cerebro-vasculares en las que ha participado Colombia. Rev Col Cardiol. 2010; 17: 195-199. [ Links ]
4. Ramsey SA, Gold ES, Aderem A. A systems biology approach to understanding atherosclerosis. EMBO Mol Med. 2010; 2: 79-89. [ Links ]
5. Grover F, Zavalza A. Endothelial dysfunction and cardiovascular risk factors. Diabetes Res Clin Pract. 2009; 84: 1-10. [ Links ]
6. Toth PP. Subclinical atherosclerosis: what it is, what it means and what we can do about it. Int J Clin Pract. 2008; 62: 1246-1254. [ Links ]
7. Dutta P, Courties G, Wei Y, Leuschner F, Gorbatov R, Robbins CS, et al. Myocardial infarction accelerates atherosclerosis. Nature. 2012; 487: 325-329. [ Links ]
8. Ghazalpour A, Doss S, Yang X, Aten J, Toomey EM, Van Nas A, et al. Thematic review series: the pathogenesis of atherosclerosis. Toward a biological network for atherosclerosis. J Lipid Res. 2004; 45: 1793-1805. [ Links ]
9. Roy H, Bhardawaj S, Yla-Herttuala S. Molecular genetics of atherosclerosis. Hum Genet. 2009; 125: 467-491. [ Links ]
10. Hadi H, Carr CS, Al Suwaidi J. Endothelial dysfunction: cardiovascular risk factors, therapy, and outcome. Vasc Health Risk Manag. 2005; 1: 183-198. [ Links ]
11. Flowers E, Froelicher E, Aouizerat BE. Gene-environment interactions in cardiovascular disease. Eur J Cardiovasc Nurs. 2012; 11: 472-478. [ Links ]
12. Elder SJ, Lichtenstein AH, Pittas AG, Roberts SB, Fuss PJ, Greenberg AS, et al. Genetic and environmental influences on factors associated with cardiovascular disease and the metabolic syndrome. J Lipid Res. 2009; 50: 1917-1926. [ Links ]
13. Epstein F, Ross R. Atherosclerosis-an inflammatory disease. The New England Journal of Medicine 1999; 340: 115-126. [ Links ]
14. Libby P, DiCarli M, Weissleder R. The vascular biology of atherosclerosis and imaging targets. J Nucl Med. 2010; 51: 33-37. [ Links ]
15. Aird WC. Endothelial cell heterogeneity. Cold Spring Harb Perspect. 2012; 2: 1-13. [ Links ]
16. Badimon L, Storey R, Vilahur G. Update on lipids, inflammation and atherothrombosis. Thromb Haemost. 2011; 105: 34-42. [ Links ]
17. Kaneto H, Katakami N, Matsuhisa M, Matsuoka TA. Role of reactive oxygen species in the progression of type 2 diabetes and atherosclerosis. Mediators Inflamm. 2010; 1-11. [ Links ]
18. Lubos E, Handy DE, Loscalzo J. Role of oxidative stress and nitric oxide in atherothrombosis. Front Biosci. 2008; 1: 5323-5344. [ Links ]
19. Sitia S, Tomasoni L, Atzeni F, Ambrosio G, Cordiano C, Catapano A, et al. From endothelial dysfunction to atherosclerosis. Autoimmun Rev. 2010; 9: 830-834. [ Links ]
20. Shao B, Heinecke JW. HDL, lipid peroxidation, and atherosclerosis. J Lipid Res. 2009; 50: 599-601. [ Links ]
21. Muller G, Goettsch C, Morawietz H. Oxidative stress and endothelial dysfunction. Hamostaseologie. 2007; 27: 5-12. [ Links ]
22. Lönn ME, Dennis JM, Stocker R. Actions of "antioxidants" in the protection against atherosclerosis. Free Radic Biol Med. 2012; 53: 863-884. [ Links ]
23. Versari D, Daghini E, Virdis A, Ghiadoni L, Taddei S. Endothelial dysfunction as a target for prevention of cardiovascular disease. Diabetes Care. 2009; 32: 314-321. [ Links ]
24. Hulsmans M, Van Dooren E, Holvoet P. Mitochondrial reactive oxygen species and risk of atherosclerosis. Curr Atheroscler Rep. 2012; 14: 264-276. [ Links ]
25. Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004; 109: III27-32. [ Links ]
26. Tousoulis D, Kampoli AM, Tentolouris C, Papageorgiou N, Stefanadis C. The role of nitric oxide on endothelial function. Curr Vasc Pharmacol. 2012; 10: 4-18. [ Links ]
27. Tabit CE, Chung WB, Hamburg NM, Vita JA. Endothelial dysfunction in diabetes mellitus: molecular mechanisms and clinical implications. Rev Endocr Metab Disord. 2010; 11: 61-74. [ Links ]
28. Bastarrachea RA, López JC, Bolado VE, Téllez J, Laviada H, Comuzzie AG. Macrófagos, inflamación, tejido adiposo, obesidad y resistencia a la insulina. Gac Méd Méx. 2007; 143: 505-512. [ Links ]
29. Deanfield JE, Halcox JP, Rabelink TJ. Endothelial function and dysfunction: testing and clinical relevance. Circulation. 2007; 115: 1285-1295. [ Links ]
30. Boyle PJ. Diabetes mellitus and macrovascular disease: mechanisms and mediators. Am J Med. 2007; 120: 12-17. [ Links ]
31. Ambrose JA, Barua RS. The pathophysiology of cigarette smoking and cardiovascular disease: An update. J Am Coll Cardiol. 2004; 43: 1731-1737. [ Links ]
32. McCully KS. Chemical pathology of homocysteine. IV. Excitotoxicity, oxidative stress, endothelial dysfunction, and inflammation. Ann Clin Lab Sci. 2009; 39: 219-232. [ Links ]
33. Sawamura T. LOX-1 a lectin-like oxidized LDL receptor identified form endothelial cells, in endothelial dysfunction. Int Congr. 2004; 1262: 531-534. [ Links ]
34. Soldatos G, Cooper ME, Jandeleit KA. Advanced-glycation end products in insulin-resistant states. Curr Hypertens Rep. 2005; 7: 96-102. [ Links ]
35. Mudau M, Genis A, Lochner A, Strijdom H. Endothelial dysfunction: the early predictor of atherosclerosis. Cardiovasc J Afr. 2012; 23: 222-231. [ Links ]
36. Avogaro A, de Kreutzenberg SV, Fadini G. Endothelial dysfunction: causes and consequences in patients with diabetes mellitus. Diabetes Res Clin Pract. 2008; 15: s94-s101. [ Links ]
37. Corrado E, Rizzo M, Coppola G, Fattouch K, Novo G, Marturana I, et al. An update on the role of markers of inflammation in atherosclerosis. J Atheroscler Thromb. 2010; 17: 1-11. [ Links ]
38. Libby P, Ridker PM, Hansson GK. Inflammation in atherosclerosis: from pathophysiology to practice. J Am Coll Cardiol. 2009; 54: 2129-2138. [ Links ]
39. Hansson GK. Inflammatory mechanisms in atherosclerosis. J Thromb Haemost. 2009; 7: 328-331. [ Links ]
40. Birukov KG. Oxidized lipids: the two faces of vascular inflammation. Curr Atheroscler Rep. 2006; 8: 223-231. [ Links ]
41. Samson S, Mundkur L, Kakkar VV. Immune response to lipoproteins in atherosclerosis. Cholesterol. 2012; 2012: 1-12. [ Links ]
42. Libby P, Okamoto Y, Rocha VZ, Folco E. Inflammation in atherosclerosis: transition from theory to practice. Circ J. 2010; 74: 213-220. [ Links ]
43. Yang H, Mohamed AS, Zhou S. Oxidized low density lipoprotein, stem cells, and atherosclerosis. Lipids Health Dis. 2012; 11: 1-9. [ Links ]
44. Ley K, Miller YI, Hedrick CC. Monocyte and macrophage dynamics during atherogenesis. Arterioscler Thromb Vasc Biol. 2011; 31: 1506-1516. [ Links ]
45. Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis. Ann Rev Immunol. 2009; 27: 165-197. [ Links ]
46. Packard RR, Libby P. Inflammation in atherosclerosis: from vascular biology to biomarker discovery and risk prediction. Clin Chem. 2008; 54: 24-38. [ Links ]
47. Keaney JF. Immune modulation of atherosclerosis. Circulation. 2011; 29: 559-560. [ Links ]
48. Zernecke A, Weber C. Chemokines in the vascular inflammatory response of atherosclerosis. Cardiovasc Res. 2010; 86: 192-201. [ Links ]
49. Johnson JL, Newby AC. Macrophage heterogeneity in atherosclerotic plaques. Curr Opin Lipidol. 2009; 20: 370-378. [ Links ]
50. Bobryshev YV. Monocyte recruitment and foam cell formation in atherosclerosis. Micron. 2006; 37: 208-222. [ Links ]
51. Poledne R, Lorenzova A, Stavek P, Valenta Z, Hubácek J, Suchánek P, et al. Proinflammatory status, genetics and atherosclerosis. Physiol Res. 2009; 58: 111-118. [ Links ]
52. Hristov M, Weber C. Differential role of monocyte subsets in atherosclerosis. Thromb Haemost. 2011; 106: 757-762. [ Links ]
53. Paoletti R, Bolego C, Poli A, Cignarella A. Metabolic syndrome, inflammation and atherosclerosis. Vasc Health Risk Manag. 2006; 2: 145-152. [ Links ]
54. Woollard KJ, Geissmann F. Monocytes in atherosclerosis: subsets and functions. Nat Rev Cardiol. 2010; 7: 77-86. [ Links ]
55. Semple JW, Freedman J. Platelets and innate immunity. Cell Mol Life Sci. 2010; 67: 499-511. [ Links ]
56. Andersson J, Libby P, Hansson GK. Adaptive immunity and atherosclerosis. Clin Immunol. 2010; 134: 33-46. [ Links ]
57. Packard RR, Lichtman AH, Libby P. Innate and adaptive immunity in atherosclerosis. Semin Immunopathol. 2009; 31: 5-22. [ Links ]
58. Boullier A, Bird DA, Chang MK, Dennis EA, Friedman P, Gillotre-Taylor K, et al. Scavenger receptors, oxidized LDL, and atherosclerosis. Ann N Y Acad Sci. 2010; 947: 214-223. [ Links ]
59. Yuan Y, Li P, Ye J. Lipid homeostasis and the formation of macrophage-derived foam cells in atherosclerosis. Protein Cell. 2012; 3: 173-181. [ Links ]
60. Ouimet M, Marcel YL. Regulation of lipid droplet cholesterol efflux from macrophage foam cells. Arterioscler Thromb Vasc Biol. 2011; 32: 575-581. [ Links ]
61. Maiti R, Agrawal NK. Atherosclerosis in diabetes mellitus: role of inflammation. Indian J Med Sci. 2007; 61: 292-306. [ Links ]
62. Libby P. Inflammation and cardiovascular disease mechanisms. Am J Clin Nutr. 2006; 83: 456-460. [ Links ]
63. Ait-Oufella H, Taleb S, Mallat Z, Tedgui A. Recent advances on the role of cytokines in atherosclerosis. Arterioscler Thromb Vasc Biol. 2011; 31: 969-979. [ Links ]
64. Douglas G, Channon KM. The pathogenesis of atherosclerosis. Medicine. 2010; 38: 397-402. [ Links ]
65. Gerthoffer WT. Mechanisms of vascular smooth muscle cell migration. Circ Res. 2007; 100: 607-621. [ Links ]
66. Raffetto JD, Khalil RA. Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem Pharmacol. 2008; 75: 346-359. [ Links ]
67. Dhawan SS, Nanjundappa A, Branch JR, Taylor WR, Quyyumi AA, Jo H, et al. Shear stress and plaque development. Expert Rev Cardiovasc Ther. 2010; 8: 545-556. [ Links ]
68. Franco M, Cooper RS, Bilal U, Fuster V. Challenges and opportunities for cardiovascular disease prevention. Am J Med. 2011; 124: 95-102. [ Links ]
69. Almeida J, Álvarez O. Fisiopatología de los síndromes coronarios agudos. Rev Cubana Med. 2006; 45: 13-24. [ Links ]
70. Borissoff JI, Spronk HM, Ten Cate H. The hemostatic system as a modulator of atherosclerosis. N Engl J Med. 2011; 364: 1746-1760. [ Links ]
71. Abbate R, Cioni G, Ricci I, Miranda M, Gori AM. Thrombosis and acute coronary syndrome. Thromb Res. 2012; 129: 235-240. [ Links ]
72. Aksu K, Donmez A, Keser G. Inflammation-induced thrombosis: mechanisms, disease associations and management. Curr Pharm Des. 2012; 18: 1478-1493. [ Links ]
73. Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. 2011; 17: 1410-1422. [ Links ]
74. Halvorsen B, Otterdal K, Dahl TB, Skjelland M, Gullestad L, Øie E, et al. Atherosclerotic plaque stability - what determines the fate of a plaque? Prog Cardiovasc Dis. 2008; 51: 183-194. [ Links ]
75. Badimon L, Padró T, Vilahur G. Atherosclerosis, platelets and thrombosis in acute ischaemic heart disease. Acute Cardiovascular Care. 2012; 1: 60-74. [ Links ]
76. Borensztajn KS, Von der Thüsen JH, Spek CA. The role of coagulation in chronic inflammatory disorders: a jack of all trades. Curr Pharm Des. 2011; 17: 9-16. [ Links ]
77. de Moerloose P, Boehlen F, Neerman-Arbez M. Fibrinogen and the risk of thrombosis. Semin Thromb Hemost. 2010; 36: 7-17. [ Links ]
78. Breitenstein A, Tanner FC, Lüscher TF. Tissue factor and cardiovascular disease: quo vadis? Circ J. 2010; 74: 3-12. [ Links ]
79. Kaplan ZS, Jackson SP. The role of platelets in atherothrombosis. Hematology Am Soc Hematol Educ Program. 2011; 51-61. [ Links ]
80. Esmon CT. The interactions between inflammation and coagulation. Br J Haematol. 2005; 131: 417-430. [ Links ]
81. Vinik AI, Erbas T, Park TS, Nolan R, Pittenger GL. Platelet dysfunction in type 2 diabetes. Diabetes Care. 2001; 24: 1476-1485. [ Links ]
82. Carr ME. Diabetes mellitus: a hypercoagulable state. J Diabetes Complications. 2001; 15: 44-54. [ Links ]
83. Jennings LK. Mechanisms of platelet activation: need for new strategies to protect against platelet-mediated atherothrombosis. Thromb Haemost. 2009; 102: 248-257. [ Links ]
84. Maguire JM, Thakkinstian A, Sturm J, Levi C, Lincz L, Parsons M, et al. Polymorphisms in platelet glycoprotein 1ba and factor VII and risk of ischemic stroke: a meta-analysis. Stroke. 2008; 39: 1710-1716. [ Links ]
85. Kleinegris M, Cate-Hoek A, Cate H. Coagulation and the vessel wall in thrombosis and atherosclerosis. Pol Arch Med Wewn. 2012; 122: 557-566. [ Links ]
86. Balagopal PB, de Ferranti SD, Cook S, Daniels SR, Gidding SS, Hayman LL, et al. Nontraditional risk factors and biomarkers for cardiovascular disease: mechanistic, research, and clinical considerations for youth. Circulation. 2011; 123: 2749-2769. [ Links ]
87. Briasoulis A, Tousoulis D, Androulakis ES, Papageorgiou N, Latsios G, Stefanadis C. Endothelial dysfunction and atherosclerosis: focus on novel therapeutic approaches. Recent Pat Cardiovasc Drug Discov. 2012; 7: 21-32. [ Links ]
88. de la Cuesta F, Barderas MG, Calvo E, Zubiri I, Maroto AS, Darde VM, et al. Secretome analysis of atherosclerotic and non-atherosclerotic arteries reveals dynamic extracellular remodeling during pathogenesis. J Proteomics. 2011; 75: 2960-2971. [ Links ]
89. Jawien J. Atherosclerosis in 2012: what is new?. Pol Arch Med Wewn. 2012; 122: 170-173. [ Links ]
90. Lenglet S, Thomas A, Chaurand P, Galan K, Mach F, Montecucco F. Molecular imaging of matrix metalloproteinases in atherosclerotic plaques. Thromb Haemost. 2012; 107: 409-416. [ Links ]
