










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Cardiología
Print version ISSN 0120-5633
Rev. Colomb. Cardiol. vol.21 no.3 Bogota May/June 2014
aUniversidad CES, Medellín, Colombia.
bClínica CES, Medellín, Colombia.
* Autor para correspondencia. Correo electrónico: mauricioduque@une.net.co (M. Duque).
Recibido el 2 de agosto de 2013; aceptado el 22 de noviembre de 2013.
El síndrome de QT corto es una entidad clínica rara, descrita hace apenas unas pocas décadas. Esto, sumado al pequeño número de casos descritos en la literatura mundial, hace difícil comprender su fisiopatología y tener una idea clara sobre el tratamiento a realizar. Hasta el momento se han descrito varias alteraciones genéticas que se asocian con síndrome de QT corto y riesgo mayor de muerte súbita, algunas que incluso lo relacionan con otros síndromes (particularmente el síndrome de Brugada). Se presenta un caso de síndrome de QT corto en quien, gracias al implante de un cardiodesfibrilador, se logró abortar un episodio de muerte súbita. En nuestro conocimiento, este es el primer caso descrito en Colombia y abre la posibilidad de que existan casos similares en el país.
Palabras clave: Muerte súbita; Arritmias; Síncope; Prevención; Desfibrilador.
Short QT syndrome is a rare clinical entity, described just a few decades ago. This, coupled with the small number of cases described in the literature, makes difficult to understand its pathophysiology and have a clear idea about the treatment to be performed. So far, several genetic alterations associated with short QT syndrome and increased risk of sudden death, some even related with other syndromes (particularly Brugada syndrome), have been described. A case of a patient with short QT syndrome in whom thanks to the implant of a defibrillator an episode of sudden death was aborted, is presented. To our knowledge, this is the first case reported in Colombia and opens the possibility of similar cases in the country.
Keywords: Sudden death; Arrhythmias; Syncope; Prevention; Defibrillator.
Introducción
En el mundo la muerte súbita es una de las entidades que mayor interés despiertan, ya que a pesar de múltiples estrategias diagnósticas, hasta ahora no hay una herramienta que permita determinar con certeza qué pacientes la desarrollarán. Hasta el momento, la mayoría de los casos se asocia con alteraciones estructurales, lo cual permite que con la evaluación morfológica y funcional del corazón se puedan establecer poblaciones con mayor riesgo. Sin embargo, hasta en el 5% al 10% de los pacientes con muerte súbita no existen alteraciones estructurales1; algunos de ellos presentan alteraciones en canales iónicos que favorecen la aparición de arritmias ventriculares malignas y muerte súbita, como son aquellos con síndrome de QT largo, síndrome de Brugada, taquicardia ventricular catecolaminérgica y síndrome de QT corto (SQTC). Sobre este último, es aún poco lo que se conoce, no sólo porque fue descrito recientemente, sino por la reducida cantidad de casos en la literatura médica mundial. A continuación se describe el que, según nuestro conocimiento, es el primer caso en Colombia.
Descripción del caso clínico
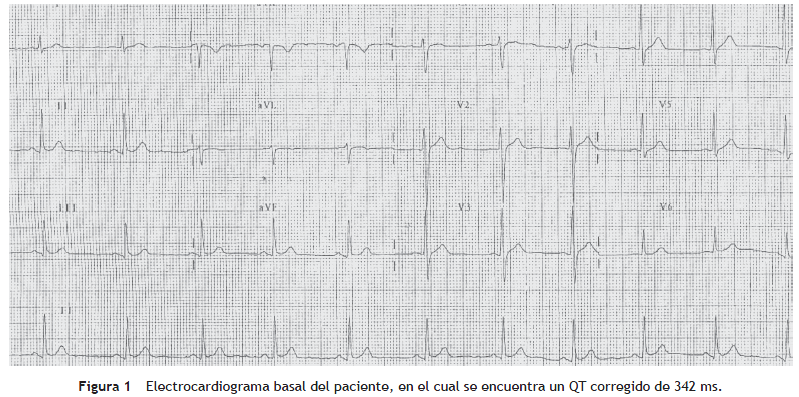
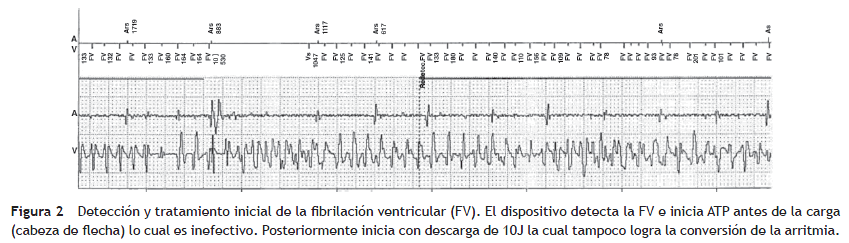
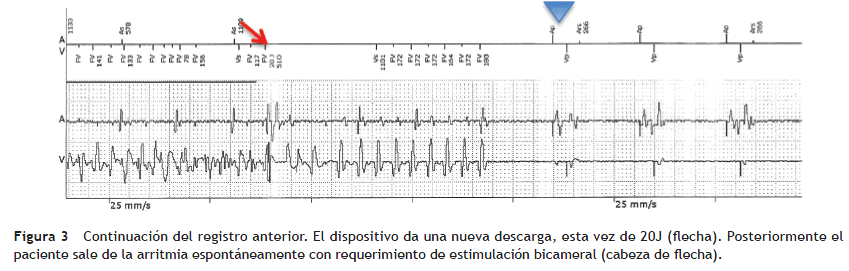
Paciente de género masculino, de 37 años, valorado en el servicio por cuadro sincopal a repetición, algunos de los cuales se presentaron en posición sentado, lo que fue interpretado como síncope de origen cardíaco. No tenía antecedentes personales de importancia ni factores de riesgo para enfermedad cardiovascular; sin embargo narró antecedente familiar de muerte súbita (el padre falleció a los 52 años; la abuela paterna a los 44 años). Mediante ecocardiografía transtorácica se documentó corazón sano en cuanto a su estructura, con un panel de química sanguínea (electrolitos y función renal) que no mostró alteraciones significativas. Al evaluar el electrocardiograma de 12 derivaciones se encontró QTc de 342 ms (fig. 1). La evaluación del resto de su familia no reveló hallazgos electrocardiográficos similares. Con el fin de estratificar el riesgo de muerte súbita, se llevó a estudio electrofisiológico, durante el cual no se lograron inducir arritmias ventriculares. Sin embargo, teniendo en cuenta los hallazgos electrocardiográficos del paciente, el antecedente de síncope y de muerte súbita familiar se le implantó un cardiodesfibrilador, el cual posteriormente demostró su utilidad al abortar un episodio de fibrilación ventricular (figs. 2 y 3).
Discusión
A principio de los años 90 se documentó una asociación entre un intervalo QT corto y el riesgo aumentado de muerte súbita en pacientes evaluados con electrocardiografía Holter. Pese a ello, fue tan sólo en el 2000 cuando se publicó la primera serie de pacientes (tres miembros de una misma familia y un paciente no relacionado que presentó un episodio de muerte súbita) que se plantearon los posibles mecanismos fisiopatológicos que podrían llegar a explicar el riesgo aumentado de muerte súbita, considerando que puede ser ocasionada por una dispersión de la repolarización (mecanismo compartido con el síndrome de Brugada)2. Desde entonces se ha despertado un interés significativo por esta rara entidad de la cual aún poco se sabe debido al pequeño número de casos reportados.
Hasta el momento se ha establecido un trasfondo genético que se asocia con un mayor o menor riesgo de muerte súbita, con mutaciones genéticas que además han sido relacionadas con fibrilación atrial3-5; incluso, estas mutaciones genéticas se asocian con fenotipos específicos. Es así como se han descrito mutaciones en los genes HERG que codifican el canal IKr1, responsable del SQTC tipo 1 (el cual se caracteriza por una onda T picuda simétrica); en el gen KCNQ1, que codifica el canal IKs y es responsable del SQTC tipo 2 (que se caracteriza por una onda T de aspecto normal); y en el gen KCNJ2 que codifica el canal rectificador de entrada IK1, el cual está asociado con el SQTC tipo 3 (que se caracteriza por una onda T asimétrica, con una fase descendente con una pendiente marcada)5-9. De manera más reciente, el grupo de Antzelevitch describió dos variantes nuevas relacionadas con mutaciones que generan pérdida de la función de los genes que codifican la subunidad a1 y b2b del canal de calcio tipo L, los cuales se asocian con un fenotipo similar al del síndrome de Brugada y que han sido descritos como SQTC tipo 4 y 5 respectivamente. La transmisión de estos genes parece ser de tipo autosómica dominante, con una alta tasa de penetración en las familias afectadas. Sin embargo, existen además casos de novo o con SQTC que no corresponden a ninguna de estas mutaciones, por lo cual no siempre existe esta relación genotipo-fenotipo. De hecho, algunos pacientes cursan tan solo con fibrilación atrial, mientras que otros presentan taquicardia ventricular polimórfica y fibrilación ventricular.
El hallazgo más característico es el QT corto en el electrocardiograma de 12 derivaciones, considerándose diagnóstico un QT menor a 340 ms o un QTc menor de 345 ms en un paciente con antecedente de fibrilación auricular o fibrilación ventricular previa. Este acortamiento es independiente de la frecuencia cardíaca (para evitar clasificar erróneamente el acortamiento del QT fisiológico que se produce con la taquicardia), y puede observarse también en el monitoreo Holter.
Estas variaciones en las manifestaciones y la genética, aunadas al pequeño número de pacientes estudiados hasta el momento, hacen que la comprensión que se tiene hasta el momento acerca del tratamiento a seguir en estos pacientes, sea muy limitada. Se acepta el uso de cardiodesfibriladores implantables aun en aquellos en quienes no es posible inducir arritmias ventriculares en el estudio electrofisiológico (se ha reportado que éste tiene una sensibilidad de apenas el 50%)10. Infortunadamente, el uso de un cardiodesfibrilador implantable plantea un problema adicional en este grupo de pacientes (particularmente con aquellos con SQTC1, que tienen una T alta) dado por el riesgo de doble conteo por parte del dispositivo (y por ende, suministrar terapias inapropiadas). En el paciente del caso, la terapia con cardiodesfibrilador demostró su utilidad al abortar un episodio de muerte súbita.
En cuanto al uso de fármacos, es poca la utilidad de los mismos. Medicamentos como la amiodarona, los beta-bloqueadores y otros grupos de antiarrítmicos no han demostrado ningún beneficio. Sin embargo, los derivados de la quinidina sí han demostrado menor riesgo de arritmias ventriculares y auriculares, prolongando el intervalo QT con disminución del riesgo de muerte súbita. Si bien aún no hay información que permita utilizar dichos tratamientos de manera rutinaria en todos los pacientes, sí se genera expectativa en la posibilidad de dar tratamiento farmacológico en el futuro.
Conclusión
Por ser un síndrome de reciente y escasa descripción, es poco lo que se sabe acerca de él. En nuestro conocimiento, este es el primer caso descrito en Colombia, lo cual permitirá tener en consideración esta entidad clínica rara y buscarla activamente en pacientes con antecedente familiar o personal de síncope o muerte súbita.
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.
Bibliografía
1. Wever EF, Robles de Medina EO. Sudden death in patients without structural heart disease. JJ Am Coll Cardiol. 2004;43: 1137-44. Epub 2004/04/06. [ Links ]
2. Gussak I, Brugada P, Brugada J, Wright RS, Kopecky SL, Chaitman BR, et al. Idiopathic short QT interval: a new clinical syndrome? Cardiology. 2000;94:99-102. Epub 2001/02/15. [ Links ]
3. Chen YH, Xu SJ, Bendahhou S, Wang XL, Wang Y, Xu WY, et al. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science (New York, NY). 2003;299:251-4. [ Links ] Epub 2003/01/11.
4. Xia M, Jin Q, Bendahhou S, He Y, Larroque MM, Chen Y, et al. A Kir2.1 gain-of-function mutation underlies familial atrial fibrillation. Biochemical and biophysical research communications. 2005;332:1012-9. Epub 2005/06/01. [ Links ]
5. Yang Y, Xia M, Jin Q, Bendahhou S, Shi J, Chen Y, et al. Identification of a KCNE2 gain-of-function mutation in patients with familial atrial fibrillation. Am J Human Genetics. 2004;75: 899-905. Epub 2004/09/16. [ Links ]
6. Brugada R, Hong K, Dumaine R, Cordeiro J, Gaita F, Borggrefe M, et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation. 2004;109:30-5. [ Links ] Epub 2003/12/17.
7. Hong K, Bjerregaard P, Gussak I, Brugada R. Short QT syndrome and atrial fibrillation caused by mutation in KCNH2. J Cardiovasc Electrophysiol. 2005;16:394-6. Epub 2005/04/15. [ Links ]
8. Hong K, Piper DR, Diaz-Valdecantos A, Brugada J, Oliva A, Burashnikov E, et al. De novo KCNQ1 mutation responsible for atrial fibrillation and short QT syndrome in utero. Cardiovascular Research. 2005;68:433-40. Epub 2005/08/20. [ Links ]
9. Priori SG, Pandit SV, Rivolta I, Berenfeld O, Ronchetti E, Dhamoon A, et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circulation research. 2005;96:800-7. Epub 2005/03/12. [ Links ]
10. Maury P, Extramiana F, Sbragia P, Giustetto C, Schimpf R, Duparc A, et al. Short QT syndrome. Update on a recent entity. Archives of cardiovascular diseases. 2008;101:779-86. Epub 2008/12/09. [ Links ]

{kind=link}
{kind=link}
{kind=link}