











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
El síndrome de deleción 22q11 consiste en una agrupación variable de características fenotípicas (ver tabla 1, tabla 2 y fig. 1), inicialmente consideradas como síndromes independientes, que tras la identificación de su origen en la microdeleción de la denominada región crítica SDG (22q11.2), se conocen como síndrome de deleción de la banda 22q11.2 (SD22q11.2)1,2.
Tabla 1 Características fenotípicas craneofaciales en SD22q11.2 (1,3,4, 6-11)
| Categoría | Características |
| Craneales | Microcefalia |
| Craneosinostosis | |
| Retrognatia o micrognatia | |
| Facies | Cara alargada |
| Asimetría facial con el llanto | |
| Oculares | Hipertelorismo |
| Fisuras palpebrales cortas | |
| Ptosis palpebral | |
| Estrabismo | |
| Ambliopía | |
| Embriotoxon posterior | |
| Vasos retinales tortuosos | |
| Vitreorretinopatía exudativa familiar | |
| Esclerocórnea | |
| Nasales | Puente nasal alto y ancho |
| Orales | Filtrum corto |
| Comisuras labiales inclinadas hacia abajo | |
| Dientes pequeños | |
| Hipodesarrollo de la cúspide lingual en los primeros premolares mandibulares | |
| Opacidad del esmalte | |
| Oídos | Orejas de baja implantación y malformadas |
| Otitis media recurrente | |
| Hipoacusia conductiva o neurosensorial | |
| Anomalías palatinas | Paladar hendido |
| Úvula bífida | |
| Paladar hendido submucoso produce voz hipernasal e incompetencia velofaríngea | |
| Labio y paladar hendidos (Extremadamente raro) | |
| Dificultades en la alimentación | Pobre succión |
| Regurgitación nasal | |
| Alteración de la deglución | |
| Cerebrales estructurales | Polimicrogiria |
| Fisura de Silvio agrandada | |
Tabla 2 Características fenotípicas no craneofaciales en SD22q11.2 (1,10,12-15,29)
| Categoría | Cracterísticas | ||
| F E N O T I P O F Í S I C O | Cardiopatías | Tetralogía de Fallot | Coartación de la aorta |
| Tronco arterioso | Comunicación interauricular (CIA) | ||
| Arco aórtico interrumpido | Estenosis pulmonar | ||
| Comunicación interventricular (CIV) | Corazón izquierdo hipoplásico | ||
| CIV + atresia pulmonar | Ductus arterioso persistente (DAP) | ||
| Otros defectos conotruncales | |||
| Endocrinológicas | Hipoparatiroidismo que causa tetanías neonatales, convulsiones y nefrocalcinosis | Deficiencia de la hormona de crecimiento con consecuente talla baja | |
| Alteraciones inmunológicas | Disfunción de Linfocitos B (LsB) | Enfermedades autoinmunes (AI) | |
| Disfunción de Linfocitos T (LsT) | Laringomalacia | ||
| Sistema respiratorio | Fístula traqueo-esofágica | Traqueomalacia | |
| Tráquea corta con disminución anillos | Broncomalacia | ||
| Cartílago tiroídeo anormal | Membrana glótica anterior congénita | ||
| Gastrointestinal | Atresia esofágica | Malrotación intestinal (MRI) | |
| Estenosis/Atresia anal | |||
| Renal y Genitourinarias | Duplicaciones renales | Agenesia renal | |
| Displasia renal multiquística | Testículos no descendidos | ||
| Hidronefrosis | Hipospadias | ||
| Esqueléticas | Escoliosis | Polidactilia | |
| Ectrodactilia | Dedos largos y cónicos | ||
| Dermatológicas | Rash cutáneo | ||
| Malignidades | Hepatoblastomas | Tumor de Wilms | |
| Carcinoma de células renales | |||
| Cognitivas, psiquiátricas y comportamentales | Retraso del desarrollo psicomotor | Déficit de atención e hiperactividad | |
| Trastorno de ansiedad | Desórdenes afectivos | ||
| Desórdenes del espectro autista | Esquizofrenia | ||
| Trastorno esquizoafectivo | |||
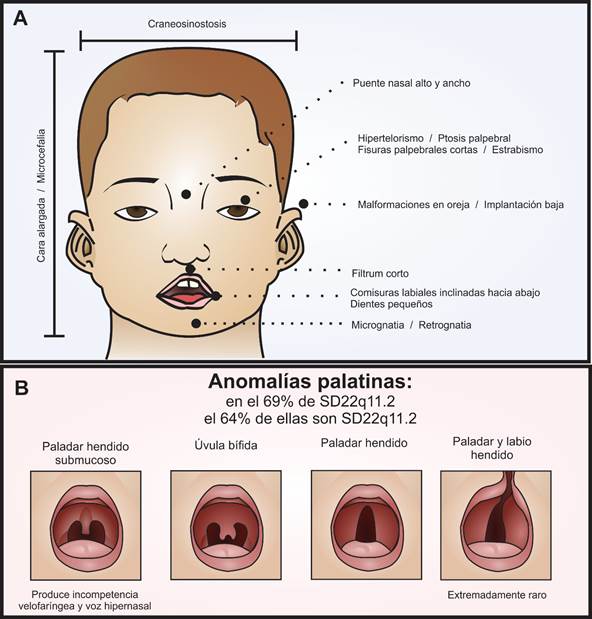
Figura 1 Fenotipo craneofacial del SD22q11.2. En el panel A se señalan las fascies características del síndrome relacionadas también en la tabla 1. En el panel B se diagraman las variantes de las anomalías palatinas en orden de compromiso de los componentes mucoso y óseo. Como grupo, las alteraciones palatinas están muy relacionadas con el síndrome, 64% de las personas con anomalías palatinas tienen SD22q11.2 y el 69% de los pacientes con SD22q11.2 tienen anomalías palatinas. Es muy raro que se presente, en este síndrome, el paladar y labio hendido.
No obstante, las características craneofaciales pueden estar ausentes o ser muy sutiles en afectados no caucásicos e infantes caucásicos menores de 10 años de edad2,3. De manera que la probabilidad de hacer un diagnóstico temprano de esta patología dependerá del conocimiento que el clínico tenga sobre las características fenotípicas no craneofaciales (ver tabla 2) que también hacen parte del síndrome1,2.
Dentro de la región crítica de deleción del síndrome de DiGeorge se encuentran varios genes, entre ellos el TBX1 (T-box 1) ha sido identificado como el mayor contribuyente al fenotipo2. Existen, sin embargo, otras causas etiológicas poco frecuentes entre las cuales se encuentran mutaciones puntuales en los genes TBX1 o en CHD7 (Chromodomain Helicase DNA Binding Protein 7) y pacientes con exposición prenatal a isotretinoina o glicemia elevada1.
La prevalencia de SD22q11.2 se ha calculado de 1 en 5.950 en la población general; 1 en 6.000 en caucásicos, afrodescendientes y asiáticos; y hasta 1 en 3.800 en hispanos. Además, 1 de cada 8 tetralogías de Fallot corresponden a esta deleción4. No obstante, la prevalencia de esta alteración difiere en gran medida entre diferentes estudios poblacionales1, razón por la cual se debe hacer un estudio independiente en cada región o entidad de salud.
El objetivo de esta publicación es realizar una revisión literaria de las bases embriológicas de las malformaciones cardiacas más comunes dentro de esta entidad sindromática, así como de los aspectos epidemiológicos, genéticos y clínicos importantes dentro del manejo de pacientes con esta patología.
Metodología
Se realizó una revisión de la literatura en inglés y español publicada en la base de datos Pubmed utilizando los siguientes términos MESH (“Genetics”, “DiGeorge Syndrome” y “22q11 Deletion Syndrome”) usando diferentes combinaciones y conectores. Se limitó la búsqueda a artículos originales en humanos y otros animales (“humans” or “animals”), revisiones literarias o sistemáticas y guías de práctica clínica, con fecha de publicación entre el año de 1980 y el 23 de febrero de 2016. Además, se incluyeron por su pertinencia y relevancia artículos encontrados en las bases de datos ScienceDirect, y SciELO. Se realizó un análisis excluyendo los artículos cuya metodología no era adecuada y se extrajo la información de los artículos elegidos a través de la producción de matrices para el manejo de la información.
Fisiopatología: genética y bases embriológicas
La cresta neural (CN) es una población celular multipotente originada en el tubo neural dorsal. Las células de la cresta neural (CCN) sufren una delaminación y migran para formar una larga lista de derivados. Algunas se convierten en células de Schwann en el sistema nervioso periférico, otras en los melanocitos, y las más craneales forman el ectomesénquima precursor de los tejidos duros y blandos de la cabeza y el cuello (hueso, dientes, parénquima y estroma de timo, paratiroides y tiroides)5. No obstante, mientras se realizaba la ablación de una porción de la CN para el estudio de la inervación parasimpática del corazón, se obtuvieron embriones sin septación aórtico-pulmonar, lo cual condujo a postular la existencia de una porción de la CN cuyas células tenían como destino final el tracto de salida del corazón, y se denominó a esta porción “cresta neural cardiaca” (CNC)6.
Posteriormente, se realizaron estudios en el ratón, usando métodos transgénicos, para marcaje genético de las CCN. Los arcos faríngeos 3, 4 y 6, que son los más caudales, contienen arterias que forman los arcos aórticos 3, 4 y 6, los cuales al remodelarse forman las grandes arterias, la arteria carótida común, el arco aórtico definitivo y el ductus arterioso7-9. La CNC se ha ubicado desde el nivel de las placodas óticas hasta el nivel del somita 3, pues a través del modelo de ablación de la CNC en el pollo, estudios retrovirales en el pollo y experimentos de la línea Cre en el ratón, se ha establecido que estas CCN se desplazan ventralmente para hacer una pausa en la cresta circunfaríngea, y luego se abren camino entre el ectodermo y el endodermo de los arcos faríngeos 3, 4 y 6, hasta llegar al tracto de salida del corazón10. Llegan a los cojines endocárdicos del tracto de salida del corazón, quienes realizan la septación aórtico-pulmonar formando la aorta y el tronco pulmonar11,12. Estos cojines han sido descritos como crestas dispuestas en espiral, que protruyen por miocardialización (invasión por miocardiocitos), y que están formadas a partir de tres poblaciones celulares mesenquimáticas diferentes.
La porción conal proximal de los cojines endocárdicos, que al protruir da origen a la masa de las valvas semilunares, se forma a partir de células endocárdicas locales que, inducidas por el miocardio conal, sufren una transformación epitelio-mesenquimática. Mientras esta masa va protruyendo, van sufriendo un remodelamiento, para dar origen a las válvulas semilunares (3 cúspides). Se han encontrado CCN en los ápices de las cúspides de válvulas en estadios temprano de su desarrollo en el ratón y el pollo12,13, pero no en las válvulas maduras del ratón8, por lo cual se plantea que las células de la CNC son reemplazadas por células mesenquimáticas (que antes eran endocárdicas)10. En el ratón, las células de la CNC tienen un papel en el establecimiento de la arquitectura valvular adecuada, posibilitando el remodelamiento valvular tardío y la apoptosis mesenquimática14.
La porción conal distal de los cojines, que al protruir ayuda a formar la parte proximal del septo aórtico-pulmonar, se forma a partir de las células mesenquimáticas provenientes de los arcos faríngeos y por las células de la CNC (sobre todo en los ratones).
La porción truncal de los cojines endocárdicos, que es la más distal, tiene forma de U invertida; ayuda a formar la parte distal del septo aórtico pulmonar en un sentido de distal a proximal, ya que sus prolongaciones protruyen de distal a proximal; y se forma a partir de células de la CNC11,15,16.
La septación aórtico-pulmonar definitiva implica que haya una desaparición del endocardio que reviste los cojines endocárdicos, que permita una mezcla de mesénquima y miocardio entre los cojines opuestos, y por tanto, su afrontamiento. Se ha propuesto un papel activo de las células de la CNC en este proceso de fusión, pues se ha encontrado la presencia de estas células en el subendocardio de la porción proximal del septo, formando una especie de costura en el septo postfusión11,12.
Se ha observado que no todas las células de la CNC llegan hasta el tracto de salida del corazón, sino que algunas se quedan en la región de los arcos aórticos, ya que si bien no son necesarias para la formación de estos arcos, sí lo son para su remodelación17; otras se quedan a ese nivel para formar el músculo liso de las arterias de los arcos18, y otras contribuyen a la formación del tracto de entrada del corazón (cerca al sistema conducente). Se sabe que las CCN proveen la totalidad de la inervación parasimpática cardiaca6, y que son los miocardiocitos los que se especializan para formar el sistema conducente cardiaco19. Hay discusión sobre si algunas de las CCN ayudan a conformar el sistema conducente cardiaco, pues aunque se han encontrado CCN en el tracto de entrada cerca del sistema conducente en los embriones del pollo y del ratón20,21, se han encontrado células negativas para Mesp-1 en el sistema His-Purkinje22; y se han encontrado CCN en el sistema conducente central13. Así que algunos consideran que no hay evidencia suficiente19. En lo que sí hay acuerdo es en que las CCN tienen un papel en el desarrollo adecuado del sistema conducente, pues en los pollos con ablación de la CNC se produce un retardo en la maduración de la función del sistema conducente13,20,21,23; y la deleción de Hf1b en la CCN produce una disfunción del receptor de neurotropina trKC y consecuentemente una disfunción de la conducción atrial y atrioventricular24. De acuerdo a los hallazgos de todos estos estudios en modelos animales, los defectos relacionados con la CNC podrían clasificarse en dos grandes grupos: el de los causados por ausencia de la contribución estructural directa de las células de la CNC, y el de los causados por alteración de la señalización y/o interacción tisular secundaria a la ausencia de las células de la CNC (ver tabla 3).
Tabla 3 Clasificación de los defectos relacionados con la cresta neural cardiaca (CNC) (32,39-42,44,46,47,49,50,58)
| Defectos relacionados con CNC | |
| Ausencia de la contribución estructural directa de las células de la CNC | Alteración de la señalización y/o interacción tisular secundaria a la ausencia de las células de la CNC |
| Defectos del septo del conontronco | Defectos de la arquitectura de las válvulas semilunares |
| Defectos del músculo liso de las arterias persistentes de los arcos aórticos (que formarán grandes arterias) | Defectos del sistema conducente cardiaco |
| Defectos del parénquima y estroma de timo, paratiroides y tiroides | Defectos del patrón adecuado de los derivados de los arcos aórtico 3, 4 y 6 |
| Defectos de la inervación parasimpática cardiaca | Defectos del alineamiento ventrículo-arterial |
Existe una población celular que contribuye a la formación del tracto de entrada y del tracto de salida cardiacos, ubicada en la faringe caudal ventral justo detrás de la unión del tracto de salida a la faringe, a la cual se le ha denominado “segundo campo cardiaco”25. La disfunción de estas células se asocia a defectos de mal alineamiento ventrículo-arterial. En embriones con ablación de la CNC, se observó que los tractos de salida no septados tenían también dextroposición, y que había una proliferación celular aumentada en el campo cardiaco secundario, lo cual llevó a plantear que la proliferación adecuada de estas células permite un alargamiento del tracto de salida y secundariamente un correcto alineamiento VA. Además, para el desarrollo normal del campo cardiaco secundario se necesita la presencia de células de la CNC26, pues estas ejercen una regulación de la actividad secretora en la faringe, modulando la expresión de FGF8 y por tanto la acción sobre su receptor, y esto a su vez permite mayor diferenciación y supervivencia celular. El manejo de modelos con deficiencia de la CCN con un bloqueador del receptor FGFR1, SU5402, Ac anti-FGF8 lleva a un rescate del plegamiento, alineamiento del tracto de salida y la función miocárdica; pero no del tronco arterioso10,27. Es decir, persiste la anomalía producida por la falta estructural de la CCN y se rescata la señalización.
El fenotipo de los embriones con ablación de la CNC incluye las malformaciones cardiacas mencionadas y alteraciones de las glándulas del cuello6,17,28,29 30. Como se ve, varias de las características fenotípicas de los embriones con ablación de la CNC se traslapan con las de los pacientes con SD22q11.2 (ver tabla 4).
Tabla 4 Fenotipo por ablación de la cresta neural cardiaca vs. fenotipo por SD22q11.2 (1,32,36,57-60)
| Hallazgos fenotípicos en la ablación de la CNC | Presencia en fenotipo por SD22q11.2 | ||
| Anomalía | Frecuencia (%) | ||
| Septación defectuosa del tracto de salida: Tronco arterioso persistente. | 90% | Sí | |
| Patrón anormal de las arterias de los arcos aórticos y las grandes arterias: Aorta interrumpida, arco aórtico doble. | 100% | Sí | |
| Plegamiento anormal del tubo cardiaco y mal alineamiento del polo arterial: ventrículo derecho de doble salida. | 10% | Sí | |
| Comunicación interventricular | ¿? | Sí | |
| Disfunción miocárdica: reducción de acoplamiento excitación-contracción, contractilidad y corriente tipo L de Ca +2 | 100% | ¿? | |
| Estenosis infundibular y de la válvula pulmonar (visto en el modelo murino PuDTK:Wnt1-Cre, pero no en el pollo) | ¿? | ¿? | |
| Alteraciones de las glándulas del cuello: hipoplasia/aplasia de timo tiroides paratiroides | 100% | Sí | |
Se sabe que la región cromosómica 22q11 está flanqueada por secuencias de ADN repetitivo, las cuales pueden llevar a entrecruzamiento no alélico durante la meiosis (espermatogénesis u ovogénesis), y por tanto, a microdeleciones. La deleción más común se encuentra en el 85% de los pacientes con SD22q11.2, tiene un tamaño de 3 millones de pares de bases y comprende aproximadamente 40 genes entre los cuales se encuentran el TBX1 (aparentemente el más correlacionado con el fenotipo), el HIRA y el UFD1L2,31.
Aspectos clínicos
Las características fenotípicas que se han descrito en pacientes con SD22q11.2 son diversas, aquí se han clasificado en dos grandes grupos: características craneofaciales y no craneofaciales (ver fig. 2 y tablas 1 y 2)2,3,31-39.
Características craneofaciales
Dentro de las características fenotípicas craneofaciales (ver fig. 1), encontramos varias posibilidades3,31-38. De estas es pertinente resaltar las anomalías palatinas, presentes en el 69% de los pacientes con SD22q11.2 y las cuales presentan una gran variabilidad clínica31,38. Los pacientes con esta alteración genética pueden presentar paladar hendido, úvula bífida, paladar hendido submucoso con incompetencia velofaríngea y voz hipernasal, y rara vez labio y paladar hendido (ver fig. 1B)3,31,35,38.
Al respecto, un 36% de los pacientes presentan dificultades en la alimentación debido a la pobre succión y regurgitación nasal secundaria a incompetencia velofaríngea, que suele mejorar al año de vida; y/o alteración de la deglución, con defectos en las fases orofaríngea y cricofaríngea2,31.
Sistema cardiaco
Podemos encontrar cardiopatías en 74-80% de los pacientes con SD22q11.22,39,31,37,40-43. Entre las identificadas, algunas suelen diagnosticarse en la infancia: tetralogía de Fallot, tronco arterioso, arco aórtico interrumpido; otras suelen diagnosticarse después de los 2 años: comunicación interventricular (CIV), atresia pulmonar con CIV, otros defectos conotruncales; y otras se han reportado solo ocasionalmente: anillo vascular, trasposición de grandes arterias con CIV, coartación de la aorta, comunicación interauricular, estenosis pulmonar, corazón izquierdo hipoplásico, ductus arterioso persistente31,44. Por ello se recomienda la realización de un electrocardiograma y un ecocardiograma a todo paciente diagnosticado con la deleción si no se ha hecho antes y valoración con cardiología pediátrica para la corrección de la anomalía si alguna malformación es evidente2.
Sistema inmunológico
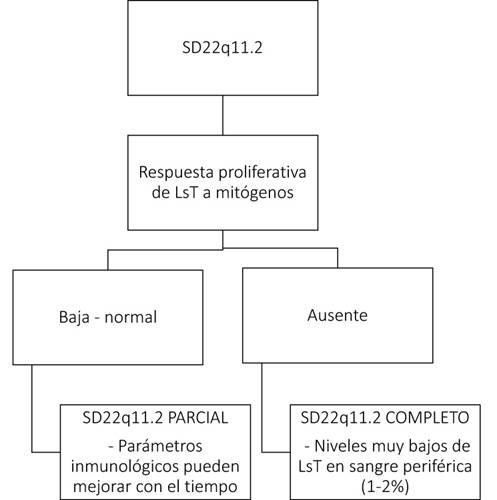
También podemos encontrar alteraciones inmunológicas en el 77% de los pacientes secundarias a hipoplasia/aplasia del timo31,45. Este hallazgo tiene su mayor relevancia entre los 3 y los 6 meses de edad, cuando los pacientes empiezan a presentar infecciones por microorganismos típicamente asociados a deficiencia de linfocitos T (LsT) como: hongos, Pneumocystis (carinii) jiroveci, e infecciones virales diseminadas46,47. Teniendo como criterio la respuesta proliferativa de LsT a mitógenos, se ha sugerido una clasificación del fenotipo inmunológico (ver fig. 3)48,49. Así, si la respuesta es baja o normal, los afectados pueden clasificarse como pacientes con SD22q11.2 parcial y sus parámetros inmunológicos pueden mejorar con el tiempo; si la respuesta es nula, los afectados pueden clasificarse como pacientes con SD22q11.2 completo con niveles muy bajos de LsT en sangre periférica48,49.
También se han descrito pacientes con hallazgos que reflejan disfunción de linfocitos B (LsB), como hipogamaglobulinemia o deficiencia selectiva de Acs (IgA), secundaria probablemente a deficiencia de LsT ayudadores2,50. Además, se ha encontrado una prevalencia aumentada de enfermedades autoinmunes (AI) como la artritis reumatoidea (AR) juvenil, púrpura trombocitopénica idiopática51,52, citopenias (Síndrome de Evans)53, uveítis AI54 y eczema severo55. Se recomienda al momento del diagnóstico un estudio del perfil inmunológico mediante conteo diferencial de leucocitos, cuantificación de las inmunoglobulinas G, A y M2. Sólo 1% de los pacientes SD22q11 no tienen linfocitos T requiriendo valoración para el transplante de Timo y profilaxis contra micosis y pneumocystis junto con la terapia de reemplazo con inmunoglobulinas2.
Hipocalcemia
El hipoparatiroidismo es otra de las características fenotípicas no craneofaciales cuya consecuencia más notable es la hipocalcemia, con tetanías y convulsiones neonatales, y la nefrocalcinosis37. Igualmente, la hipocalcemia se presenta en 17-60% de los pacientes con SD22q11.2, no obstante, el 50% de los afectados no requieren suplementación al año de edad31,44. Se sabe que aunque tengan niveles de calcio y hormona paratiroidea normales, en los pacientes con SD22q11.2 usualmente la reserva secretora de esta hormona está disminuida56.
Fenotipos de alta probabilidad e indicaciones para buscar la deleción
El fenotipo de alta probabilidad varía ligeramente a medida que el paciente avanza en su ciclo vital (ver tabla 5)31,38. Se han establecido fenotipos puntuales como indicaciones para buscar la deleción, definidos mediante la facilidad de su hallazgo en cada etapa del ciclo vital56. En la etapa neonatal comúnmente se presentan: las cardiopatías conotruncales, el paladar hendido y la hipocalcemia. Por el contrario durante la niñez temprana es común la deficiencia inmune severa. Los pacientes con SD22q11.2 pueden presentar a cualquier edad 2 o más de las siguientes: las cardiopatías conotruncales, los defectos palatinos, la voz hipernasal, el reflujo nasofaríngeo, la discapacidad del aprendizaje/RDSM, los problemas psiquiátricos/comportamentales, la inmunodeficiencia, la hipocalcemia y las facies típicas31,38.
Tabla 5 Fenotipos de alta probabilidad para SD22q11.2 (11,12)
| Característica clínica | Frecuencia en pacientes con SD22q11.2 (%) | Importancia en etapa neonatal | Importancia en el 1º año de vida | Importancia en el 2º y 3º años de vida | Importancia en la etapa escolar |
| Dificultad del aprendizaje/retraso del desarrollo | 70-90 | + | + | ++ | |
| Cardiopatía | 74-80 | + | + | + | + |
| Inmunodeficiencia | 77 | + | ++ | + | + |
| Anomalía palatina | 69 | + | + | + | + |
| Hipocalcemia | 17-60 | ++ | + | ||
| Dificultad para la alimentación | 36 | + | + | ||
Igualmente, se ha calculado qué proporción de pacientes con ciertas anomalías específicas tienen deleción en 22q, haciéndose recomendaciones sobre si se debe o no buscar la deleción ante la presencia de dichas anomalías (ver tabla 6)31,56.
Tabla 6 Riesgo de deleción en 22q en situaciones clínicas específicas
| Anomalía | Qué proporción son SD22q11.2? (%) | Buscar SD22q11.2? |
| Cardiopatía conotruncal aislada | 20-30 | Sí |
| Hipocalcemia neonatal | 74 | Sí |
| Arco aórtico interrumpido | 50-60 | Sí |
| Insuficiencia velofaríngea | 64 | Sí |
| Cualquier cardiopatía detectada tardíamente | 1 | No |
| Esquizofrenia | 0-6 | No |
Diagnóstico
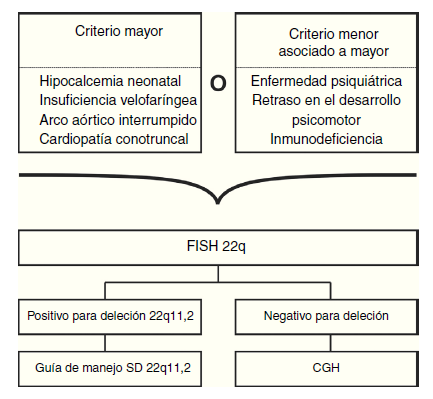
Ante la variabilidad genética de un fenotipo sugestivo de SD22q11.2, la hibridación genómica comparativa con microarreglos (CGH por el inglés: comparative genomic hybridization array) es la prueba más apropiada, pues además, de detectar deleciones en 22q11.2, es capaz de detectar otras deleciones/duplicaciones, tanto grandes como submicroscópicas, y además, aunque por el momento no se ha demostrado ninguna correlación clínica de acuerdo al tamaño de la deleción, el CGH provee información detallada sobre los puntos de corte57,58.
Si el CGH no es costeable, o no está disponible, la primera opción es solicitar un FISH para 22q11.2 con un cariotipo (ver fig. 4)2,31,59, una segunda opción es solicitar el cariotipo y el Multiplex ligation-dependent probe amplification (MLPA), pues parece que esta última prueba es equivalente al FISH, y puede realizarse en ADN extraído de gotas de sangre de papel filtro2,31,59.
Reflexión de los autores
El SD22q11.2 tiene una incidencia de 1/2000-1/4000, pero se considera que dos grandes problemas son: el subdiagnóstico y el diagnóstico tardío.
El SD22q11.2 es una causa frecuente de paladar hendido asociado a cardiopatía.
Entre los pacientes con SD22q11.2, los que tienen mayor probabilidad de diagnóstico neonatal son los que tienen cardiopatías severas e hipocalcemia.
Se han establecido fenotipos muy puntuales de cada etapa del ciclo vital como indicaciones para buscar la deleción: “cardiopatía conotruncal + paladar hendido”, “hipocalcemia + paladar hendido”, “deficiencia inmune severa”, etcétera.
Todo paciente con cualquiera de las siguientes anomalías debería ser evaluado para deleción en 22q11.2:
Cardiopatía conotruncal aislada.
Hipocalcemia neonatal.
Arco aórtico interrumpido.
Insuficiencia velofaríngea.
- Varias de las características fenotípicas de los embriones con ablación de la CNC se traslapan con las de los pacientes con SD22q11.2.
- Si el CGH no es costeable, o no está disponible, las opciones son solicitar un FISH para 22q11.2 con un cariotipo, o solicitar MLPA y cariotipo.
- Existen guías de manejo para SD22q11.2, que son el producto de consensos internacionales.

