











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIsquemia miocárdica irreversible
Tipos de muerte celular (necrosis, apoptosis, autofagia)
La muerte celular de los cardiomiocitos puede estar mediada por 3 procesos citológicos diferentes llamados: la necrosis, la apoptosis y la autofagia. La muerte irreversible es más comúnmente mediada por la necrosis, una pérdida permanente de la viabilidad dada por los cambios isquémicos irreparables. Tradicionalmente la triada de: la necrosis, la apoptosis y la autofagia controlan la muerte celular y la perduración de las vías que generan estos procesos, que son distintas y diferentes. La necrosis es una muerte celular no controlada, accidental y regulada eficientemente por la pérdida celular intrínseca para mantener o guardar la viabilidad celular. De otro lado, la apoptosis es una muerte celular programada bien controlada, por mecanismos moleculares tanto extrínsecos como intrínsecos. Un nuevo concepto muy interesante para la muerte celular es la autofagia, que es un mecanismo de sobrevida celular, inicialmente mediado por la degradación y reciclaje de organelas celulares que cuando fracasa lleva a un exceso en la degradación celular y desencadena un mecanismo de muerte celular. En efecto, algunos estudios muestran daño de los cardiomiocitos con características de autofagia durante la falla cardiaca1.
Necrosis
La necrosis es frecuentemente definida como una muerte no controlada, carente de características encontradas en la muerte celular programada2. Esta muestra características como: la ruptura de la membrana plasmática y la dilatación de las organelas citoplasmáticas, especialmente, la mitocondria3,4. Las mitocondrias son la fuente para la producción de energía, la homeostasis del Ca2+ y finalmente, la muerte celular. La permeabilidad transicional de la membrana mitocondrial, conocida como despolarización mitocondrial, se define como la pérdida del potencial transmembrana de la membrana interna de la mitocondria regulada por el incremento del Ca2+, del cual depende la permeabilidad de la membrana mitocondrial5,6. Además , la permeabilidad transicional de la membrana es inducida por la translocación del nucleótido de adenina 42 CypD, familia de la ciclofilina y las moléculas transisomerasa7. La falta de regulación en la permeabilidad transicional de la membrana resulta en la pérdida del gradiente de los protones y acaba con la producción del ATP de la fosforilación oxidativa, llevando a la tumefacción mitocondrial y ruptura de la membrana externa. Los canales de permeabilidad transicional de la membrana mitocondrial también abren sus poros a través de la membrana interna mitocondrial para estimular el incremento del Ca2+, el fosfato inorgánico, el pH alcalino y las especies reactivas de oxígeno. Estas especies reactivas de oxígeno han mostrado potenciar la muerte celular necrótica en patologías isquémicas del miocardio como la isquemia de reperfusión al lesionar el cardiomiocito8,9. El estrés oxidativo, además, induce un aumento de necrosis abriendo el poro de permeabilidad transicional de la membrana y disminuyendo la cantidad de ATP10,11. Curiosamente, la deficiencia de CypD en modelos de ratones muestra una reducción en el tamaño del infarto y resistencia a la isquemia de reperfusión inducida por la lesión del cardiomiocito8.
Apoptosis
La apoptosis se caracteriza por un proceso bien conocido de la muerte celular, programado o guiado por los mediadores celulares, los moleculares y los bioquímicos. Los eventos celulares muestran cambios característicos a nivel nuclear y citoplasmático incluyendo: condensación y fragmentación de la cromatina, cambios menores en los organelos citoplasmáticos, contracción celular, protrusión de la membrana plasmática y formación de cuerpos apoptóticos12. Procesos moleculares y bioquímicos son mediados por las vías intrínsecas y extrínsecas. En la vía intrínseca, las moléculas de proteínas de la familia BCL2 que promueven la apoptosis, aumentan la permeabilidad en la membrana externa mitocondrial llevando a un incremento en la conducción del citocromo C dentro del espacio intermembrana del citoplasma; este se une con la proteasa apoptótica activadora del factor I en presencia de ATP llevando a la formación del <<apoptosoma>> 1. Esto genera la fragmentación de la procaspasa-9, formando la caspasa-3 activa, la cual causa la muerte celular13.
En la vía extrínseca, el ligando de la muerte celular FAS ligando es activado junto con el factor de necrosis tumoral alfa y se une con los receptores de la muerte intracelular afines en la membrana plasmática. Este proceso inicia la liberación de la caspasa-8 activa a través de proteínas adaptadoras, lo que genera regulación decreciente de la vía, debido a la inducción de la caspasa-3 activa sin la mediación de la proteína BCL214,15.
La apoptosis es muy rara en los cardiomiocitos normales con una relación de 1:10.000-100.00016. Al contrario, la apoptosis está notablemente incrementada y juega un rol central en la fisiopatología de la progresión de enfermedades como la cardiomiopatía, el infarto del miocardio, la hipertrofia cardiaca y la falla cardiaca17-19. La quinasa 1 reguladora de señal de la apoptosis (ASK1) es una proteína-quinasa mitogénica activada por radicales superóxido3, y se ha visto en la sobrecarga de presión y en los corazones de ratones postinfarto20.
Autofagia
La autofagia es un proceso de la sobrevida celular en células desprovistas de nutrientes, por el reciclaje intrínseco de las organelas citoplasmáticas21,22. En la autofagia celular las proteínas propias citosólicas y organelas son secuestradas para formar "autofagosomas". Estos autofagosomas se degradan en los lisosomas bajo el control de la degradación enzimática del proteosoma de ubiquitina. La autofagia es vista tanto en la sobrevida celular como en la muerte23,24. En los corazones humanos enfermos la autofagia se observa en la hipertrofia del miocardio25, en la falla miocárdica causada por la cardiomiopatía dilatada26,27, la enfermedad valvular cardiaca28, y la enfermedad cardiaca isquémica.
Los miocitos pueden experimentar un complejo intercambio de: la necrosis, la apoptosis y la autofagia dependiendo del contexto de energía, estatus nutricional e inflamación asociada. Cuando hay un buen balance de energía el proceso de la apoptosis continúa; no obstante, si la energía disminuye por debajo de un nivel mínimo y se asocia con inflamación, las células cambian a la vía de la necrosis. En el contexto de la lesión crónica de los miocitos, estos pueden eludir la autofagia al inicio de la muerte del miocito. Por tanto, el complejo contexto del tiempo y espacio de una lesión irreversible puede modular la triada de la necrosis, la apoptosis y la autofagia, y la relativa influencia de cada mecanismo en el infarto del miocardio es controversial29.
Isquemia miocárdica reversible
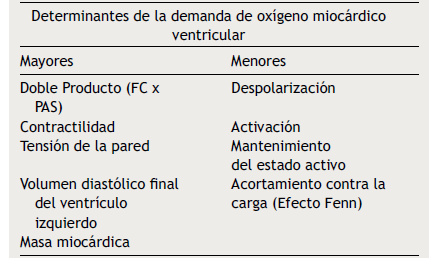
La isquemia miocárdica reversible puede ser isquemia de demanda, cuando el requerimiento de oxígeno de las células del miocardio aumenta (tabla 1), o isquemia de suplencia cuando hay obstrucción de uno de los vasos epicárdicos. Cualquier incremento en la demanda de oxígeno del miocardio debe ser equilibrada con un incremento en el flujo. La extracción del oxígeno por las células es máximo en las condiciones de reposo basal. El no balance en la demanda y la suplencia desencadena la isquemia. Si se mantiene, la isquemia juega una serie de cambios bioquímicos dados por cambios físicos del miocardio, conocido esto como la cascada isquémica (fig. 1). El flujo sanguíneo miocárdico es un complejo juego entre la permeabilidad epicárdica, microvascular, y el metabolismo del miocito25. El flujo coronario en reposo se mantiene hasta que la estenosis alcanza un 90%. Solo después de una estenosis del 95% este flujo no puede satisfacer los requerimientos de oxígeno y la angina ocurre en reposo. En lesiones del 50% o menos de estenosis, la demanda de oxígeno miocárdico se alcanza tanto en reposo como en vasodilatación máxima. No obstante, el flujo sanguíneo coronario a la máxima vasodilatación cae por debajo del nivel isquémico en estenosis del 75% o más. Esto todavía puede modificarse disminuyendo el requerimiento de oxígeno del miocardio, el cual es el objetivo de la terapia médica.
Tabla 1 Determinantes mayores y menores de la demanda de oxígeno miocárdico ventricular. FC: Frecuencia cardiaca. PAS: Presión arterial sistólica
Isquemia aguda
La isquemia aguda puede ser dada por una placa ateroesclerótica no obstructiva con superposición de espasmo y trombosis o una placa ateroesclerótica obstructiva con o sin inflamación sistémica30.
Las consecuencias de la isquemia aguda dependen de la severidad de la oclusión, la cantidad de miocardio suplida por el vaso y la presencia de circulación colateral. Cuando el flujo de reserva cae por debajo del valor autorregulatorio la isquemia subendocárdica inicia y los cambios electrocardiográficos pueden ser vistos dentro de dos minutos. No obstante, en algunos pacientes esta condición es transitoria y reversible antes del desarrollo del dolor torácico. En la isquemia silente, los pacientes pueden tener numerosos episodios con diferentes efectos en el miocardio dependiendo de la duración y la estenosis (fig. 2). Como se mencionó en la primera etapa del artículo, la isquemia aguda es totalmente reversible; sin embargo, si persiste puede llevar a la muerte del miocito. La isquemia intermitente puede preservar el músculo miocárdico disminuyendo el consumo de oxígeno y permitiendo la sobrevida del miocardio incluso en la oclusión completa del vaso. Este mecanismo de defensa se conoce como preacondicionamiento.
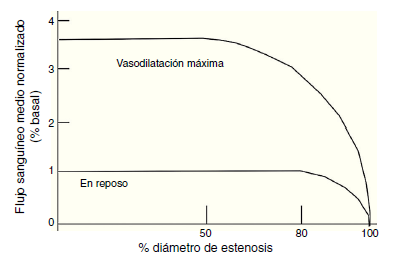
Figura 2 Esta gráfica ilustra la relación entre el flujo sanguíneo máximo y en reposo a diferentes grados de estenosis coronaria. Bajo condiciones normales la estenosis hasta del 40% aproximadamente no va alterar el flujo sanguíneo, por lo que el flujo de reserva va a mantenerse normal. Entre el 40% y 80% de estenosis, existe un flujo sanguíneo miocárdico normal en reposo, pero el flujo sanguíneo estará disminuido en la vasodilatación máxima.
Preacondicionamiento
El preacondicionamiento isquémico, se refiere a la protección impartida al miocardio por episodios previos subletales de isquemia. Originalmente descrita por Murry y Col., en 1986, el preacondicionamiento isquémico fue demostrado en el animal experimental canino luego de cuatro episodios de oclusión coronaria cada uno separado por 10 minutos de reperfusión, produciendo la misma pérdida del ATP y daño miocárdico al final de los 4 episodios, que 10 minutos simples de isquemia. No obstante, 40 minutos de isquemia fueron asociados con severa disminución de ATP y muerte celular. Este preacondicionamiento miocárdico protege contra 40 minutos de isquemia continúa pero no contra 3 horas. Por tanto, se demostró que el efecto de preacondicionamiento agudo se atenúa en aproximadamente entre 1 y 3 horas. Esta protección temprana es llamada preacondicionamiento isquémico agudo. No obstante, existe otro período de preacondicionamiento isquémico que aparece a las 12-24 horas y es llamado preacondicionamiento tardío, o segunda ventana del preacondicionamiento31. (fig. 3).
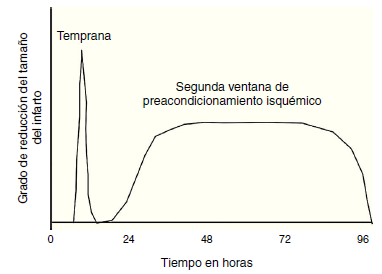
Figura 3 Representación de la naturaleza temporal de las dos ventanas de preacondicionamiento. Esta muestra que la ventana inicial de protección es transitoria, la forma tardía de protección reaparece dentro de las 24 horas del estímulo de preacondicionamiento, al que se le llama segunda ventana de protección. Esta a pesar de no tener tanto grado de protección, tiene un período más prolongado, entre 12 y 72 horas luego del estímulo de preacondicionamiento.
El preacondicionamiento isquémico es una respuesta gradual y depende de la duración de la isquemia. Experimentos en animales muestran que la isquemia de menos de 2 minutos de duración y más de 10 minutos, seguida por la reperfusión no confieren ninguna protección28. Sin embargo, en humanos, el rol del preacondicionamiento isquémico es visto en escenarios clínicos como el ejercicio, la angioplastia y los puentes coronarios. "La angina de calentamiento" ocurre cuando el paciente es capaz de caminar más después de reposar un primer episodio de dolor anginoso32. Durante la angioplastia, las inflaciones subsecuentes luego de la primera inflación se asocian con: menores cambios electrocardiográficos, de dolor, producción de lactato y liberación de enzimas; en parte dependiente de la presencia de circulación colateral33. Pacientes con angina previa al infarto tienen pequeños infartos, impidiendo un mayor deterioro de la función del ventrículo izquierdo, con reducción de la incidencia de la falla cardiaca, el choque y la mortalidad34. El pinzamiento transitorio de la aorta antes de un pinzamiento prolongado ofrece la protección durante la cirugía de puentes arteriales coronarios35. El preacondicionamiento puede ocurrir tanto en la isquemia de demanda como en la de suplencia. El preacondicionamiento isquémico agudo causa disminución del 50-80% del tamaño del infarto mientras el preacondicionamiento isquémico tardío lo reduce en un 30-40%. Esto también reduce las arritmias, los cambios del ST y la severidad de la angina. Sin embargo, todos los estudios han mostrado que el preacondicionamiento isquémico puede retardar la muerte celular pero no prevenirla del todo si la revascularización no se hace luego de ciertas horas.
Mecanismos de preacondicionamiento
Preacondicionamiento agudo
El desarrollo del preacondicionamiento agudo involucra varios mecanismos que incluyen: el desencadenamiento o iniciación por un estímulo, las vías de transducción de señal que actúan como mediadores, los canales de KATP que funcionan como los efectores y los efectos finales36. Durante un pequeño período isquémico, el corazón parece liberar: la adenosina, las bradiquininas, la norepinefrina y los opioides que disparan el preacondicionamiento isquémico. Los mediadores previos a través de sus respectivos receptores proteína G activados activan la proteína quinasa C y se iniciará una cascada de reacciones llevando al preacondicionamiento isquémico. Una vez el preacondicionamiento isquémico comienza no tiene reversión por el bloqueo de la proteína quinasa C sugiriendo un efecto de memoria37. Los radicales libres también desencadenan el preacondicionamiento por activación directa de las proteínas quinasa protectoras. Otras sustancias y características como: el calcio, la longitud de la fibras miocárdicas, el período transitorio de hipertermia, la tirosín quinasa y otras quinasas mitogénicas activadas también se ha visto que juegan un rol en el preacondicionamiento.
La fig. 4 ilustra un ejemplo de como múltiples receptores actúan en paralelo en el preacondicionamiento isquémico. En el primer grupo, 5 minutos de isquemia alcanza el umbral de protección, pero en el segundo grupo, 3 minutos de isquemia no lo alcanzan. En el tercer grupo, bloqueando los receptores B2 de las bradiquininas con HOE 140 (potente inhibidor de las bradiquininas de larga acción), causa que los 5 minutos de isquemia no sean protectores por que estos ya no pueden alcanzar el umbral. Al contrario, el aumento de las bradiquininas contribuye con el inhibidor de la enzima convertidora de angiotensina, que previene la degradación
de las mismas, permitiendo 3 minutos de isquemia para alcanzar un umbral protector38. Los canales de KATP juegan un importante rol en el acondicionamiento isquémico. Estos son llamados ATP sensibles por que están cerrados con niveles fisiológicos de ATP. Los canales de KATP son encontrados tanto en la sarcómera como en la mitocondria; no obstante, son los canales de KATP de la mitocondria quienes juegan un rol más importante en el preacondicionamiento isquémico. Hay varias teorías que soportan el beneficio de la apertura de los canales de KATP mitocondrial. Una de las teorías más aceptadas es que la apertura de los canales de K causa la entrada de K dentro de la mitocondria, previene la entrada de calcio durante la lesión y por tanto, previene la muerte celular. Esto también causa mayor fosforilación de la creatina que actúa como suministro de energía al citoplasma durante los episodios de isquemia adicionales.
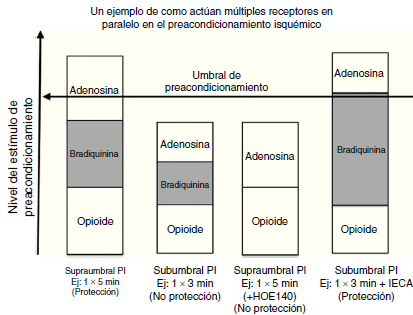
Figura 4 Un ejemplo de cómo múltiples receptores actúan en paralelo en el preacondicionamiento isquémico. En el primer panel, 5 minutos de isquemia alcanza el umbral de protección, pero en el segundo panel, 3 minutos de isquemia no lo alcanza. En el tercer panel, bloqueando las bradiquininas del receptor B2 con HOE 140 va a causar que un período de 5 minutos de isquemia se vuelva no protector al no alcanzar el umbral. Al aumentar las bradiquininas contribuyendo con el IECA que previene la degradación de estas, permite que 3 minutos de isquemia alcancen el umbral protector. Modificada Circ Res. 1995 Sep;77(3):611-21(39).
Preacondicionamiento tardío
El preacondicionamiento tardío aparece alrededor de las 2 horas y puede durar hasta 72 horas. Este es más débil que el preacondicionamiento primario. Algunos desencadenantes envueltos en el preacondicionamiento temprano también juegan un papel importante en el tardío, como la adenosina por vía de los receptores A1 y A3, el óxido nítrico, los radicales libres y los canales de potasio ATP mitocondriales36.
Implicaciones clínicas del preacondicionamiento
Los síndromes coronarios agudos en humanos pueden presentarse con o sin elevación del segmento ST, la angina inestable y la muerte súbita coronaria. Pacientes que presentan un síndrome coronario agudo con elevación del segmento ST, tienen una oclusión total en sus arterias y necesitan una reperfusión urgente, ya sea con la trombólisis o con la intervención coronaria percutánea. El preacondicionamiento isquémico no juega un rol en este tipo de pacientes. Sin embargo, en pacientes con síndrome coronario agudo sin elevación del segmento ST o angina inestable, caracterizados por episodios previos de oclusión y reperfusión, el preacondicionamiento isquémico puede ser beneficioso. Varias condiciones clínicas en las cuales el preacondicionamiento isquémico ocurre pueden ayudarnos a encontrar formas terapéuticas para inducir el mismo. No obstante, se debe recordar que el intervalo de tiempo entre el inicio de los síntomas y la revascularización es más importante y no se puede demorar esperando el preacondicionamiento.
El reto real es ¿cómo va a hacer el preacondicionamiento para no retardar el tiempo de reperfusión? Esto genera la necesidad de intentar reproducir el preacondicionamiento generando el mismo efecto pero sin causar angina; con medicamentos como: la adenosina, los agonistas de adenosina, los agonistas de proteína quinasa C, los estimuladores de apertura de canales de KATP y los imitadores del óxido nítrico que preacondicionan a nivel celular39. El nicorandil es un estimulador de apertura de los canales de KATP aprobado en Europa y Japón como medicamento antianginoso40. La adenosina ha probado ser beneficiosa en pacientes con infarto de miocardio. Un estudio reciente REOPEN-AMI mostró que la adenosina intracoronaria alivia la obstrucción microvascular al compararla con el nitroprusiato de sodio intracoronario. La infusión de la adenosina administrada como terapia de reperfusión dentro de las tres horas de un síndrome coronario agudo con elevación del segmento ST aumenta la sobrevida temprana y tardía y reduce la muerte y la falla cardiaca a los 6 meses. Esta no tiene efecto en pacientes más allá de las 3 horas del síndrome coronario agudo con elevación del segmento ST. Esto puede ser porque el efecto benéfico del preacondicionamiento no está presente en la isquemia prolongada41. Sin embargo, este rol del preacondicionamiento luego de la reperfusión no se conoce de manera clara. Aún no hay claridad en el beneficio del preacondicionamiento en los pacientes mayores, los diabéticos y con la falla cardiaca.
Posacondicionamiento
La reperfusión es el tratamiento definitivo para la isquemia miocárdica pero en algunos pacientes esta viene con un alto precio por la lesión de la reperfusión caracterizada por el daño mediado por: los radicales libres, el daño endotelial, la inflamación celular, la alteración intracelular de la homeostasis del calcio y la muerte celular. El posacondicionamiento se refiere al proceso protector del miocardio de la lesión por reperfusión. Staat y Cols., mostraron que cuatro ciclos de 1 minuto de oclusión con balón seguido por 1 minuto de reperfusión, luego de un implante directo de stent en un síndrome coronario agudo, comparado solamente con el implante del stent se asoció con menor liberación de CK-MB, mejor grado de Blush miocárdico, mejor resolución del segmento ST y menor fenómeno de no reflujo sin aumento de las complicaciones42. El posacondicionamiento parece compartir el mecanismo molecular de preacondicionamiento. Este promueve la función de poros mitocondriales, por tanto, previene el daño osmótico a la célula. El concepto de posacondicionamiento parece ser más explicable en la práctica; ciclos de la isquemia y la reperfusión pueden hacerse en el laboratorio de cateterismo con angioplastia con balón, pero el beneficio de la duración de la isquemia inducida por la angioplastia en el posacondicionamiento es desconocida. La cardioprotección profiláctica parece ser promisoria para el daño isquémico usando nuevos agentes que pueden activar vías de supervivencia a través del preacondicionamiento y del posacondicionamiento.
Miocardio aturdido
Se ha visto que luego de una lesión isquémica reversible la contractilidad miocárdica permanece deprimida a pesar del restablecimiento de un flujo normal de sangre. Este fenómeno de disfunción mecánica temporal luego de una lesión isquémica pero con flujo sanguíneo normal y en ausencia de cualquier lesión irreversible es llamado miocardio aturdido. Este, es un estado completamente reversible y responde a catecolaminas. La severidad de la disfunción miocárdica es determinada por la duración y la severidad de la isquemia43. Se sabe que la estenosis entre el 40-50% de los vasos epicárdicos no limita el flujo sanguíneo en reposo ni en ejercicio. La estenosis entre el 50-90% no limita el flujo sanguíneo en reposo pero puede desarrollar isquemia cuando la demanda aumenta. Estos episodios de isquemia intermitente pueden causar aturdimiento. Así mismo, cuando la oclusión coronaria es de menos de 20 minutos y el flujo se restablece, el miocardio desarrolla aturdimiento. Largos períodos de oclusión pueden causar áreas de infarto y aturdimiento intercaladas. Los efectos de la isquemia y la reperfusión en el corazón están basados en estudios de modelos caninos anestesiados con oclusiones de las arterias coronarias proximales. Períodos cortos de isquemia menores de 20 minutos seguidos por la reperfusión no son asociados con el desarrollo de la necrosis y pueden resultar en fenómenos de aturdimiento y preacondicionamiento. Si la duración de la oclusión coronaria se extiende más allá de 20 minutos, un frente de onda de necrosis se dirige del subendocardio al subepicardio. La reperfusión antes de 3 horas de isquemia salva el tejido isquémico viable (este tejido salvado puede presentar aturdimiento). La reperfusión más allá de 3 a 6 horas en este modelo no reduce el tamaño del infarto. La reperfusión tardía aún puede tener efectos benéficos en la reducción y prevención de la expansión del infarto del miocardio y el remodelado ventricular izquierdo. Clínicamente, el aturdimiento puede ocurrir seguido del ejercicio en presencia de: una estenosis que limita el flujo, una angina vasoespástica en combinación con isquemia miocárdica, el infarto del miocardio con reperfusión temprana o seguido de isquemia global de cirugía de puentes arteriales coronarios. El miocardio aturdido no resulta en anormalidades electrocardiográficas; este es una falta de balance entre el flujo y la función. Varios mecanismos son descritos para el aturdimiento, (tabla 2) pero el más aceptado es la lesión mediada por los radicales libres como resultado de la reperfusión. El anión superóxido, el peróxido de hidrogeno y el ion hidroxilo son radicales libres responsables de la mayoría de los daños. Estas reacciones son canalizadas por el hierro (reacción de Fenton) y la xantina oxidasa. Boli y cols., han mostrado que tras la infusión de GMP la antioxidación atenuaba el aturdimiento si se comienza antes de la reperfusión o dentro del primer minuto de la reperfusión, pero no tiene efecto si se inicia luego del primer minuto44. Este experimento mostró que si se inhibe la producción de radicales libres durante la explosión inicial, el aturdimiento puede ser mitigado. Otros agentes como la desferoxamina y el captopril han sido usados para reciclar los radicales libres en los ensayos clínicos.

Miocardio hibernado
El miocardio hibernado se define como una región miocárdica viable, sin contractilidad. Esto obedece a una reducción severa del flujo sanguíneo miocárdico pero insuficiente para generar muerte celular. El consumo del poco oxígeno es utilizado, exclusivamente, para la supervivencia celular, más insuficiente para generar la contractilidad. La definición se acompaña de mejoría de la función contráctil después de la revascularización45. La hibernación miocárdica aparece como una extensión del miocardio aturdido. Al inicio el flujo sanguíneo es normal y la función deprimida, lentamente el flujo sanguíneo disminuye y el miocardio progresa de un corto estado de hibernación a una hibernación prolongada. El período de tiempo de la mejoría de la función contráctil luego de la revascularización depende de la duración y la severidad de la estenosis. El miocardio hibernante ha sido documentado en: angina, infarto del miocardio, aneurisma del ventrículo izquierdo, muerte súbita cardiaca abortada y enfermedad valvular con pobre función del ventrículo izquierdo. Estudios experimentales han mostrado que la enfermedad de un vaso puede llevar a cambios de hibernación en áreas remotas46. Muchos pacientes con miocardio hibernante presentan disfunción del ventrículo izquierdo más que síntomas de isquemia. Disminución del flujo sanguíneo miocárdico, especialmente, el flujo sanguíneo miocárdico subendocárdico es la alteración fundamental en la hibernación crónica. Durante el proceso de hibernación, varios cambios estructurales afectan al miocito, a la microcirculación y a la matriz extracelular.
Daño del miocito
La hibernación puede ser considerada como respuesta adaptativa a la disminución del flujo sanguíneo miocárdico. La reducción crónica del suministro de sangre causa apoptosis del miocito, y hay una pérdida regional del 30% de los miocitos durante la transición del miocardio crónicamente aturdido a hibernante47. Como resultado de la apoptosis, los miocitos restantes van a hipertrofiarse para mantener el grosor. Habrá también pérdida de los miofilamentos y las sarcómeras dentro de la célula. El espacio vacío es ocupado por partículas de glicógeno. Las mitocondrias aumentan en número pero su tamaño disminuye y, por tanto, son llamadas minimitocondrias. El retículo sarcoplásmico y los túbulos en T son raramente vistos y la conexina-43 y las uniones GAP se subexpresan. Existe la evidencia de que una regulación creciente del mecanismo cardioprotector en respuesta a la isquemia repetitiva, y una regulación decreciente de la sintasa-quinasa de glucógeno 3 B puede aumentar la sobrevida celular48. Por otro lado, otros estudios que involucran biopsias humanas han mostrado una regulación creciente de proteínas proapoptóticas que llevan al incremento de la muerte celular y la fibrosis49. La heterogeneidad de los estudios es probablemente dada por la diferencia de la duración de la isquemia en diversas poblaciones de pacientes.
Matriz extracelular y microcirculación
La matriz extracelular se caracteriza por un incremento de colágeno tipo I, III y depósitos de fibronectina. Estudios han mostrado que la mejoría luego de la revascularización fue mayor en áreas donde los capilares estaban intactos y menor en los lugares donde existía fibrosis y disyunción de la arquitectura capilar.
Metabolismo
Los miocitos hibernantes alteran su metabolismo como adaptación a la disminución del flujo sanguíneo y el contenido de glicógeno aumenta. No obstante, el fosfato de creatina y los niveles de ATP no están alterados, contrastando con el aturdimiento en donde el ATP está disminuido. Hay una regulación decreciente de múltiples proteínas envueltas en el metabolismo oxidativo y transporte de los electrones48. El miocardio hibernante es también caracterizado por un desorden de los receptores adrenérgicos y la presencia de fibrosis intersticial. La falta de homogeneidad en los receptores simpáticos genera un alto riesgo de las arritmias y la muerte súbita cardiaca. Algunos estudios han mostrado que la cantidad de fibras intersticiales afectan los desenlaces luego de la revascularización miocárdica. Un miocardio con gran cantidad de fibrosis > 30% está destinado a una muerte irreversible de los miocitos. La cantidad de fibrosis depende de la duración del miocardio hibernante49.

