











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
Las cardiopatías familiares hacen referencia a aquellas enfermedades cardiovasculares de causa genética, y que en consecuencia, pueden tener una presentación familiar. Entre estas se incluyen miocardiopatías, canalopatías, enfermedades de dilatación aórtica y otros síndromes hereditarios que afectan al sistema cardiovascular1,2. Aunque tienen alta variabilidad clínica, comparten como característica en común su asociación con muerte súbita, que en algunos casos puede ser la primera o única manifestación clínica2. Por estas razones, es importante establecer programas enfocados hacia la valoración clínica y genética tanto del paciente como de sus familiares, con el fin de identificar portadores asintomáticos que se puedan beneficiar de medidas preventivas para eventos cardiovasculares. Dada la alta heterogeneidad genética de estas enfermedades, múltiples variantes en distintos genes pueden generar fenotipos clínicos similares, por tanto es necesario llevar a cabo un estudio genético completo que permita identificar los defectos genéticos causales1. Con la implementación de las plataformas comerciales de secuenciación masiva o Next Generation Sequencing, hoy en día es posible llevar a cabo dicho estudio mediante un único test1,2. Aunque estas nuevas plataformas hacen posible secuenciar el genoma de un individuo a partir de una cantidad mínima de ADN extraído de sangre o saliva, solo un adecuado análisis e interpretación permitirán llegar a un diagnóstico preciso.
Este artículo pretende realizar una revisión de la arquitectura genómica de las cardiopatías heredables y proporcionar un enfoque práctico acerca de la utilidad de las tecnologías de secuenciación y de la gestión de la información como herramientas fundamentales en el diagnóstico y la mejor aproximación pronóstica de estas patologías. Este nuevo enfoque de la medicina busca identificar y comprender la predisposición/resistencia genética del individuo para desarrollar enfermedades e implica un nuevo modelo de medicina: predictiva, de precisión y personalizada.
Gestión adecuada de la información en la era genómica
La secuenciación masiva permite analizar gran cantidad de ADN a un costo cada vez más razonable, convirtiéndose en una herramienta clínica altamente costo-eficiente para el estudio de estas enfermedades. Esta tecnología está permitiendo la identificación de cientos de variantes patogénicas y asociaciones causales con el desarrollo de enfermedad, ampliando nuestro conocimiento acerca de las correlaciones genotipo-fenotipo y explorando mecanismos alternativos de patogenicidad3,4.
Estas plataformas de secuenciación requieren una serie de procesos comunes: fragmentación del ADN, purificación, enriquecimiento, obtención de librerías, secuenciación y análisis bioinformático3 (fig. 1). La información obtenida, limitada por la complejidad de la arquitectura genética y la gran cantidad de datos generados, debe ser almacenada de la mejor manera para el análisis adecuado y la gestión e interpretación precisa. Por ello se requiere mayor cantidad de recursos humanos y tecnológicos con el fin de garantizar la calidad de los resultados. Otra consecuencia de este aumento de datos es que cada vez es mayor el número de variantes de significado clínico incierto detectadas en cada estudio4. Aún no se dispone de un conocimiento suficiente para entender por completo la arquitectura genética de las cardiopatías heredables y de sus variantes asociadas.
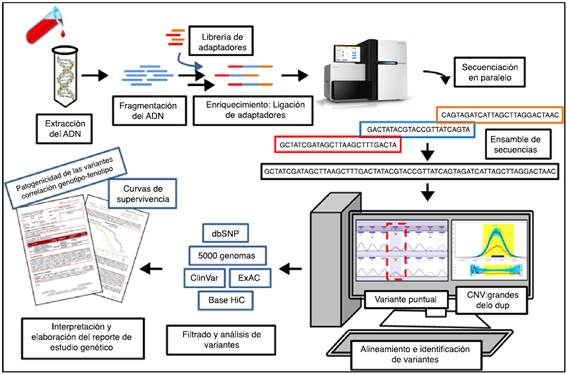
Figura 1 Proceso de secuenciación del ADN mediante secuenciación masiva (NGS) y gestión de la información. De una muestra de ADN se puede secuenciar parte o todo el genoma en un mismo ensayo. Este tipo de estudio genera una gran cantidad de información que requiere un análisis exhaustivo por un equipo multidisciplinar con el fin de lograr una adecuada interpretación de los datos. Este último proceso es importante para la elaboración de informes genéticos que sean de utilidad en la práctica clínica.
Finalmente, la secuenciación masiva puede estar dirigida a una porción del genoma, como es el caso de exoma, o a un conjunto predefinido de genes de interés como serían los paneles específicos de genes. Dada la sensibilidad de los secuenciadores de nueva generación, cada fragmento debe ser leído varias veces y tener cobertura óptima para garantizar la calidad y la fiabilidad de la secuencia analizada. En este sentido, la secuenciación de paneles de genes dirigidos ofrece una mejor cobertura de las regiones de interés que la secuenciación de porciones más grandes del genoma, aspecto crucial para garantizar un análisis completo y adecuado. A su vez, cuanto mayor número de regiones analizadas, mayor número de datos que requieren ser almacenados y analizados adecuadamente; aumentando la posibilidad de identificar variantes de significado incierto o variantes clínicamente relevantes relacionadas con enfermedades que no eran objeto de este estudio (hallazgos incidentales).
Por todo lo anterior es necesario disponer de una plataforma tecnológica de almacenamiento de datos y de un equipo multidisciplinario especializado que realice el análisis de los mismos integrando la información básica y clínica, enfocando su tarea a identificar asociaciones genotipo-fenotipo con un alto nivel de evidencia clínica.
Genética clínica de las cardiopatías familiares
Las cardiopatías familiares pueden presentar diferentes patrones de herencia: autosómicas dominantes o recesivas, ligadas al X y de patrón matrilineal o mitocondrial. Este aspecto puede ayudar a orientar el diagnóstico, pero dado que alteraciones en múltiples genes se pueden asociar al desarrollo de una misma enfermedad, su diagnóstico clínico correcto no siempre es fácil2,5. En algunos casos la presencia de genotipos complejos, de acuerdo con el número de genes implicados o de factores ambientales como el deporte competitivo o la edad, influyen en la expresión de la enfermedad5. En este panorama, genes diferentes pueden dar lugar a una expresión clínica similar (un fenotipo similar), pero también un mismo gen puede causar varios fenotipos distintos6. Incluso una misma mutación puede ser causa de una expresividad variable del fenotipo7,8 (fig. 2B). A toda esta variabilidad contribuyen factores genéticos, epigenéticos y ambientales adicionales parcialmente comprendidos y no siempre identificables (fig. 2A).
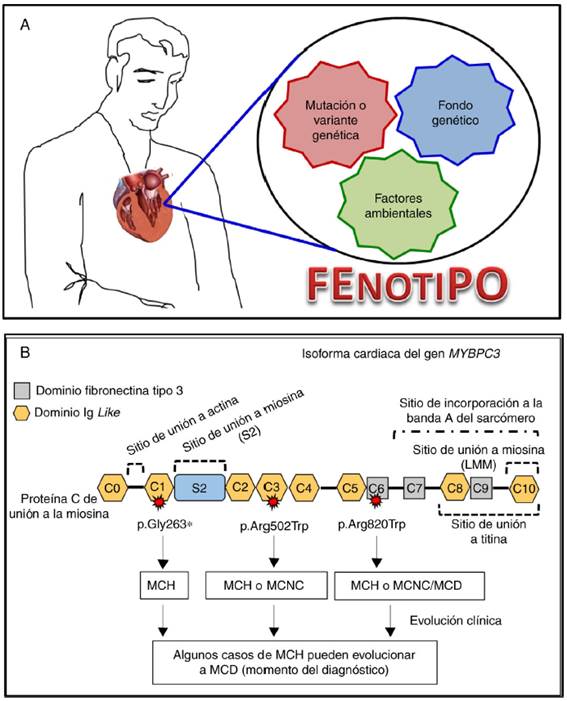
Figura 2 Correlación clínico-molecular de variantes patogénicas en las miocardiopatías. A. La expresión clínica de la enfermedad es el resultado de la interacción de una mutación con factores genéticos (presencia de segundas mutaciones en el mismo u otros genes) y/o ambientales (deporte, miocarditis, entre otros) adicionales. B. Diferentes variantes patogénicas en un gen sarcomérico (se pone de ejemplo al gen MYBPC3 que codifica la isoforma cardiaca de proteína C de unión a la miosina) pueden asociarse a diferentes tipos de miocardiopatías. A su vez, una misma variante puede dar lugar a diferentes fenotipos, incluso entre portadores de una misma familia. MCH: miocardiopatía hipertrófica; MCD: miocardiopatía dilatada; MCNC: miocardiopatía no compactada.
Por otra parte, no siempre todos los individuos portadores de variantes patogénicas desarrollarán enfermedad, y en ocasiones la aparición de una sola variante puede no ser causa suficiente para su expresión.
¿Por qué hacer un estudio genético y cómo realizar la valoración clínica-genética en la familia?
Existen recomendaciones específicas para la realización de un estudio genético en cada grupo de estas enfermedades que se revisarán más adelante. Sin embargo, las indicaciones generales podrán resumirse en las siguientes:
Para realizar un diagnóstico etiológico adecuado9,10. La identificación causal de la enfermedad es indispensable para el establecimiento de una terapia médica propicia y una mejor intervención preventiva de eventos cardiovasculares.
Para facilitar el screening familiar. En el caso de enfermedades heredables los familiares deben someterse a evaluaciones periódicas con la consiguiente carga asistencial y psicológica que esto conlleva. Conocer la variante o variantes patogénicas de la enfermedad en el caso índice de la familia permite identificar a los portadores, y tranquilizar a aquellos que no lo son, finalizando su seguimiento9,10.
Como herramienta de mejor aproximación pronóstica. Las cardiopatías familiares son clínica y genéticamente heterogéneas. La estratificación pronóstica es una parte esencial de la evaluación de este grupo de pacientes. El conocimiento de una variante patogénica permite realizar un pronóstico asociado a ésta, constituyéndose en un elemento adicional en la estratificación del riesgo, complementando los estudios clínicos tradicionales (fig. 3)11.
Para dar consejo genético. El asesoramiento genético comprende el proceso de explicar las consecuencias y naturaleza de la enfermedad, las probabilidades de desarrollarla y transmitirla, y las opciones de tratamiento y planificación familiar para las parejas con deseo de futura descendencia12.
Como herramienta de investigación clínica. Estas enfermedades aún están parcialmente comprendidas, por lo cual no se dispone de información clínica suficiente, estratificación pronóstica, y en muchos casos, ni de terapias médicas adecuadas. El mejor entendimiento de la etiología, el curso natural y el pronóstico de cada una de estas patologías permitirá avanzar en estos aspectos.
Para identificar casos beneficiarios de un tratamiento específico. Existe un subgrupo de enfermedades asociadas con el desarrollo de la miocardiopatía hipertrófica, como es la enfermedad de Fabry, que en la actualidad cuenta con un tratamiento específico13. El diagnóstico de estas entidades en la mayoría de los casos es de tipo molecular, en cuyo caso el estudio genético permite hacer un diagnóstico diferencial temprano.
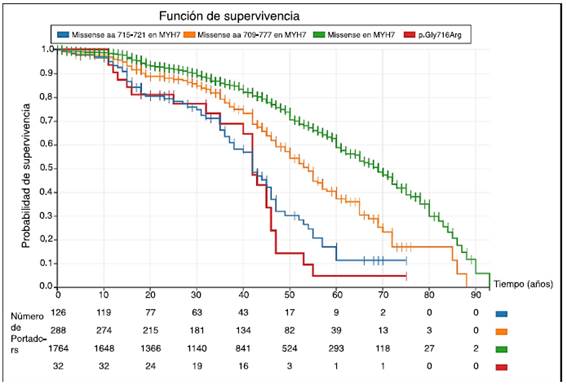
Figura 3 Curvas de sobrevida en portadores de variantes tipo missense en el gen MYH7 (Kaplan-Meier). En las curvas se observa una mayor incidencia de eventos cardiovasculares en los portadores de las variantes de tipo missense localizadas entre los aminoácidos 715 a 721 (incluyendo la variante p.Gly716Arg) al compararlas con las variantes del mismo tipo en el resto del gen 9. Este gráfico permite comprender varios aspectos relevantes: primero, no todas variantes del mismo tipo descritas en un gen no tienen por qué tener un comportamiento clínico similar; segundo, el pronóstico de los portadores puede variar según la localización de la variante dentro del gen; y tercero, la caracterización clínica de los portadores permite identificar regiones críticas del gen que ayudan a establecer una mejor valoración del pronóstico en sus portadores. Los eventos cardiovasculares fueron definidos como: muerte súbita cardiaca, muerte por insuficiencia cardiaca o trasplante, muerte por accidente cerebrovascular, relacionada con intervención cardiovascular o por otras causas cardiovasculares, descarga apropiada del desfibrilador.
La valoración clínica debe ser el primer paso antes de realizar un estudio genético en una familia. Los protocolos de las unidades de cardiopatías familiares sugieren una valoración cardiológica completa, incluyendo ECG y ecocardiograma, principalmente en los miembros de primer grado del individuo afectado y hasta donde sea posible2. La historia clínica debe incluir una buena anamnesis, antecedentes personales y familiares, número de afectados y grado de parentesco, hallazgos positivos en el examen físico e identificación de otras alteraciones físicas que pueden ser cruciales para orientar el diagnóstico. En este punto, realizar en la consulta un árbol genealógico permitirá detallar de forma fácil y sencilla los datos familiares clínicamente relevantes. Estos árboles familiares deben incluir datos de al menos tres a cuatro generaciones, ampliando el árbol en la rama familiar, materna o paterna, donde se identifiquen otros miembros afectados. El estudio genético inicialmente solo está indicado en el caso índice de la familia (individuo afectado), previo consentimiento informado. Siempre debe explicarse al paciente sobre sus beneficios, consecuencias y limitaciones. Los resultados, tanto positivos como negativos, deben ser explicados por un especialista experto en estas patologías. Una vez identificados los miembros en riesgo y la causa genética en el caso índice, se debe intentar evaluar de manera organizada a los familiares del caso índice para hacer el estudio de cribado genético o de cosegregación de la variante con la enfermedad2,9.
¿Cómo realizar la valoración de la patogenicidad de una variante genética y cómo interpretar el resultado de un estudio genético?
La interpretación de los resultados de un estudio genético tiene grandes repercusiones en la valoración clínica-genética tanto del individuo afectado como de su familia. La asignación cuidadosa de la patogenicidad de una variante es un aspecto clínicamente relevante en los reportes de los resultados de un estudio genético (tabla 1)14. Para la valoración de la patogenicidad de una variante genética se deben tener en cuenta varios aspectos: si la variante se encuentra reportada en la literatura asociada a enfermedad, si está demostrada su cosegregación con una enfermedad específica, su frecuencia alélica en la población general, el tipo de variante, su localización a nivel del gen/proteína, el grado de conservación del aminoácido afectado y su impacto sobre las propiedades fisicoquímicas de la proteína y los resultados de estudios funcionales existentes y/o de los predictores bioinformáticos1,14. Es importante recalcar que la mayoría de estas variables son solo datos orientativos a tener en cuenta dentro de esta valoración y que el aspecto más importante como evidencia de patogenicidad es la confirmación de su cosegregación con la enfermedad.
Tabla 1 Clasificación de las variantes de acuerdo con la información disponible
| Clasificación de la variante | Utilidad clínica |
| Patogénica Criterios mayores (Variantes previamente descritas asociadas a enfermedad con un alto nivel de evidencia clínica y consenso acerca de su patogenicidad. (Cosegregación demostrada con un fenotipo específico: identificación en al menos cinco probandos (casos índice) o cosegregación en al menos dos familias distintas. (Variante de tipo truncamiento (tipo frameshift, nonsense o que afecta a los sitios canónicos de splicing [+1, +2, -1, -2]) en un gen donde la pérdida de función ha sido demostrada claramente como mecanismo de patogenicidad y que cumpla al menos un criterio de apoyo. Criterios de apoyo a. Variante de tipo truncamiento (tipo frameshift, nonsense o que afecta al splicing) en un gen donde la pérdida de función es relevante. b. Presentación de novo (padres no afectos y no portadores de la variante). c. Estudios funcionales que sugieren un efecto deletéreo o no tolerable. d. Variante missense que genera el mismo cambio de aminoácido que una variante previamente descrita como patogénica. e. No identificada en población control o con muy baja frecuencia alélica a nivel poblacional (MAF<0,001%) | Valor clínico predictivo de enfermedad Para consejo genético Recomendado estudio familiar Diagnóstico prenatal o preimplantacional |
| Muy posiblemente patogénica Criterios mayores (Variante de tipo truncamiento en un gen donde la pérdida de función es relevante y que cumpla al menos un criterio de apoyo. (Variante missense o deleción in frame (que no altera el marco de lectura) en zona no repetitiva del gen con correlación genotipo-demostrada y que cumpla al menos 2 criterios de apoyo. Criterios de apoyo a. Variante localizada en una región donde se han descrito variantes asociadas con enfermedad. b. Presentación de novo c. Cosegregación con la enfermedad o presente en varios casos índices con el mismo fenotipo clínico, pero sin cumplir criterios para ser patogénica. d. Estudios funcionales que sugieren que podrían ser patogénica. e. No identificada en población control o con muy baja frecuencia alélica (MAF<0,001%) | Valor clínico predictivo de enfermedad Consejo genético Recomendado estudio familiar |
| Posiblemente patogénica Criterios mayores (Variante de tipo truncamiento en un gen donde la pérdida de función no se ha comprobado como mecanismo patogénico o no cumple criterios para considerarla patogénica. (Variante intrónica fuera de los sitios relevantes para el proceso de corte y empalme del ARN (splicing) pero cuyo análisis bioinformático sugiere que afectaría este proceso. (Variante missense o deleción in frame en zona no repetitiva del gen que no cumple criterios para ser patogénica pero que cumple con tres criterios de apoyo. Criterios de apoyo a. Identificada en población control con muy baja frecuencia alélica (MAF<0,01%). b. Presentación de novo asociada a un nuevo fenotipo clínico en la familia. c. Estudio clínico y genético del paciente y familiares sugieren una correlación de la variante con la enfermedad. d. Localizada en un dominio o región relevante del gen en el que previamente se han descrito variantes patogénicas. e. Descrita en al menos dos individuos no relacionados con el mismo fenotipo clínico. f. Estudios bioinformáticos coinciden en que tendría un efecto deletéreo. | Por el momento sin valor clínico predictivo de enfermedad La evaluación de cosegregación puede ser útil para definir su patogenicidad |
| Patogenicidad incierta o de significado clínico incierto Criterios (Variantes con información contradictoria respecto a su patogenicidad (no se ha confirmado cosegregación con la enfermedad; no hay estudios funcionales o estos no son concluyentes) (No cumplen criterios para ser incluidas en otra categoría de la clasificación. | Por el momento sin valor clínico predictivo de enfermedad La evaluación de cosegregación si hay familiares afectados y sólo en el contexto de investigación |
| Posiblemente no patogénica o benigna Criterios mayores (La frecuencia alélica de la variante en población control es mayor que la esperada para la enfermedad, o presenta una alta frecuencia alélica poblacional (> 0,05% pero ≤ 0,5%). (Está descrita en individuos con fenotipos no relacionados. (No cosegregación con la enfermedad en al menos un familiar. (Cumple al menos dos criterios de apoyo. Criterios de apoyo Variante de tipo missense en un gen en donde sólo las variantes de tipo truncamiento han demostrado ser causa de enfermedad. Estudio funcional que demuestre que la variante no altera la estructura o función de la proteína que codifica. Predictores bioinformáticos coincidentes en que la variante no alteraría la función de la proteína (se incluyen las variantes intrónicas fuera de la zona relevante de splicing). Inserciones/deleciones in frame en una región repetitiva del gen sin función conocida. Presencia de la variante en homocigosis en población control. | Sin valor clínico predictivo de enfermedad No recomendado incluirlas en el estudio genético familiar |
| No patogénica o benigna Criterios mayores (Identificada en población control con una frecuencia alélica muy alta (similar a la de un polimorfismo >0,5-1%) o con alta evidencia clínica acerca de su no patogenicidad. (No presenta cosegregación con la enfermedad en al menos dos familias. (Cumple al menos cuatro de los criterios de apoyo. Criterios de apoyo Frecuencia alélica mayor que la esperada para la enfermedad, o MAF >0,05%. Ausencia de cosegregación de la variante con el fenotipo clínico en al menos un familiar. Estudio funcional que demuestre que la variante no altera la estructura o la función de la proteína que codifica. Presencia de la variante en individuos sanos (no afectados) a una edad en la cual la enfermedad debería haberse expresado. | Sin valor clínico predictivo de enfermedad No incluirlas en el estudio genético familiar |
Modificada de Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
Cuanto más rara es una variante genética, menor es su frecuencia alélica en la población general y mayor es la probabilidad de que pueda asociarse a enfermedad. De la misma manera, es poco probable que las variantes genéticas consideradas polimorfismos (frecuencias alélicas en población general > 0,5%) sean causales de enfermedad en forma aislada (fig. 4). En general, se considera que las variantes ausentes o con frecuencias muy bajas en población general, aquellas reportadas previamente en la literatura asociadas con el desarrollo de enfermedad, o que afectan regiones funcionalmente relevantes a nivel de proteína, tienen mayor probabilidad de ser patogénicas. En la tabla 1 se presenta una clasificación de las diferentes categorías en las que se puede enmarcar un hallazgo de un estudio genético, en tanto que en la figura 4 se muestra una correlación entre la frecuencia poblacional de una variante y su posible patogenicidad.
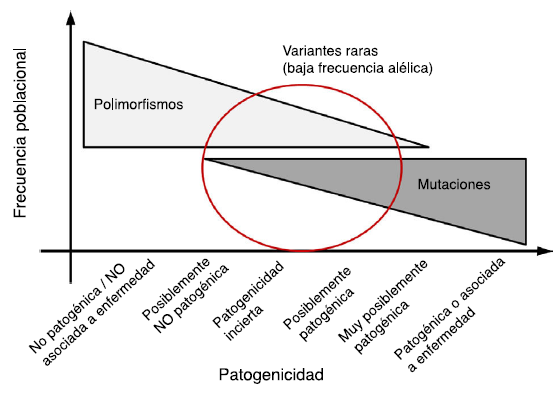
Figura 4 Relación entre la frecuencia de una variante y su posible patogenicidad. En general las variantes con alta frecuencia alélica poblacional (polimorfismo) tienen baja probabilidad de ser patogénicas; mientras que variantes no presentes en la población general tienen alta probabilidad de ser patogénicas. Sin embargo, en la parte media del espectro existe un solapamiento entre variantes raras (con baja frecuencia alélica) que pueden ser causa de enfermedad o por el contrario no ser patogénicas.
Por consenso, si hay dudas en la patogenicidad de la variante genética, “de significado clínico incierto”, no se recomienda su utilización con valor clínico predictivo de enfermedad1,9,14. Es decir, que hasta no tener más información no debería ser utilizada para la toma de decisiones clínicas en sus portadores. En algunos casos seleccionados es recomendable realizar el cribado familiar y evaluar la cosegregación de la variante con el fenotipo clínico, y determinar si solo está presente en individuos afectados o también en sanos, en un contexto de investigación. Si estas variantes son reclasificadas como patogénicas, pueden ser utilizadas con fines clínicos predictivos de enfermedad. Es importante remarcar que ser portador de una variante patogénica no implica estar enfermo, sino presentar una mayor predisposición que la población general a desarrollar una enfermedad específica, razón por la cual se requiere un seguimiento adecuado.
En general, los estudios genéticos que permiten identificar variantes patogénicas o probablemente patogénicas son considerados resultados informativos, ya que podrían modificar el manejo médico del paciente y sus familiares identificados como portadores. Por el contrario, aquellos estudios que no identifican variantes clínicamente relevantes son considerados como resultados no informativos, ya que no permiten cambiar el manejo médico del paciente, ni identificar familiares en riesgo de desarrollar la enfermedad, y en este caso se considera a todos sus miembros población en riesgo. La identificación de variantes de patogenicidad incierta debe considerarse como un resultado no informativo, con la salvedad de que en aquellos casos en los que se establezca su patogenicidad, se demuestre su cosegregación con la enfermedad o sea posteriormente descrita como claramente asociada a enfermedad, pasaría a ser un resultado informativo.
Cardiopatías familiares
Miocardiopatías
La Sociedad Europea de Cardiología define a las miocardiopatías como alteraciones miocárdicas en las cuales el músculo cardiaco está estructural y/o funcionalmente alterado, en ausencia de factores predisponentes, enfermedad coronaria, hipertensión, enfermedad valvular o enfermedad cardiaca congénita, que pueda explicar dichas anomalías15. Pertenecen a este grupo las miocardiopatías hipertrófica, dilatada, arritmogénica, restrictiva y la no compactada9. En este apartado se comentarán particularmente las tres primeras, por ser las más frecuentes. Para consultar datos acerca de la genética de las otras miocardiopatías se recomienda al lector referirse a los documentos de consenso de las Sociedades Europea y Americana de Cardiología16,17.
La miocardiopatía hipertrófica de define como la presencia de hipertrofia ventricular izquierda en ausencia de factores hemodinámicos que la expliquen15,17. Es una enfermedad genética, con un patrón de herencia autosómico dominante. Se estima que la prevalencia de esta enfermedad es de 1:500 adultos17,18. De acuerdo con datos norteamericanos, la miocardiopatía hipertrófica es responsable de la mayor parte de las muertes súbitas que se producen en sujetos jóvenes, antes de 35 años de edad, aparentemente sanos, y particularmente en la subpoblación de atletas. La incidencia anual de muerte súbita en pacientes con miocardiopatía hipertrófica varía entre 0,1 y 1,7%; se acepta como regla general que es <1%, con subgrupos de pacientes de alto riesgo en donde la misma podría ser mayor19.
Entre el 50-70% de los pacientes con miocardiopatía hipertrófica son portadores de al menos una variante patogénica en un gen sarcomérico, siendo algo mayor este porcentaje en las formas que cumplen criterios clínicos y de presentación familiar, y disminuyendo en aquellos que presentan formas esporádicas o cuando el diagnóstico no es claro20,21. Los genes más implicados en el desarrollo de miocardiopatía hipertrófica son MYH7 y MYBPC3, que codifican para la cadena pesada de la beta miosina y la proteína C de fijación a la miosina, respectivamente, ya que explican hasta el 50% de los casos20,22. Desde 1990 se han descrito más de 1.500 mutaciones asociadas al desarrollo de miocardiopatía hipertrófica en múltiples genes, particularmente en genes sarcoméricos. Sin embargo, otros genes no sarcoméricos se han asociado progresivamente con el desarrollo de la enfermedad (tabla 2). También es importante tener en cuenta la presencia de fenocopias de la miocardiopatía hipertrófica (fenotipos clínicos similares), que en su mayoría se trata de enfermedades de depósito, enfermedades de Fabry, Pompe y Danon, déficit de PRKAG2, cuyo diagnóstico diferencial en muchos casos solo es posible mediante el estudio genético.
Tabla 2 Genes relacionados con el desarrollo de las diferentes miocardiopatías y rendimiento del test genético en estas entidades. 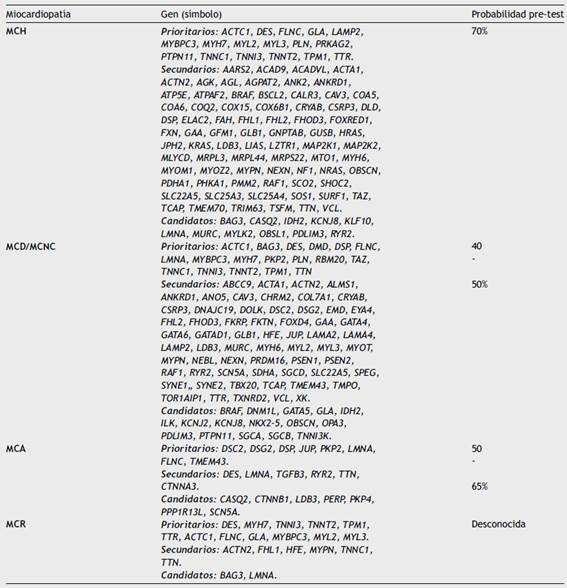
En las guías de actuación de la Sociedad Europea de Cardiología se recomienda el estudio genético en todos los pacientes con diagnóstico de miocardiopatía hipertrófica y familiares en riesgo (nivel de evidencia Ib)23.
La miocardiopatía arritmogénica se caracteriza por el reemplazo del tejido miocárdico ventricular por tejido fibroso y adiposo, asociado a un riesgo elevado de arritmias ventriculares. Desde el punto de vista epidemiológico, tiene una prevalencia de entre 1:2.000 a 1:5000 individuos de población general24. Su diagnóstico clínico se basa en una serie de criterios arrítmicos, electrocardiográficos, morfológicos y familiares, pero suele ser complejo ya que la expresión de la enfermedad muestra gran variabilidad. Tradicionalmente se acepta que el mecanismo de herencia es autosómico dominante, y en algunos casos la sola presencia de una variante es suficiente para que el fenotipo se exprese; algunas pueden asociarse a un mal pronóstico. Sin embargo, no es infrecuente que una variante genética considerada patogénica requiera de factores adicionales genéticos, presencia de segundas variantes, o ambientales, deporte competitivo, infecciones virales, etc., para producir enfermedad25. También existen formas recesivas de miocardiopatía arritmogénica caracterizadas por afectación extracardiaca (de piel y faneras), como es el caso de la enfermedad de Naxos y Carvajal25. En general, aproximadamente el 60% de los pacientes con diagnóstico de miocardiopatía arritmogénica son portadores de una variante patogénica causal identificable26.
La miocardiopatía arritmogénica se ha asociado principalmente con mutaciones en los genes desmosomales (DSC2, DSG2, DSP, JUP, PKP2 (tabla 2). El gen comúnmente identificado es el PKP2, seguido de la DSP y DSG2. Como se mencionaba previamente, es frecuente que exista más de una variante genética causal en una familia afectada y que esta combinación explique el desarrollo de la enfermedad. Una excepción son las variantes patogénicas en el gen DSP, y en especial las variantes de tipo ”radical” que generarían truncamientos a nivel de la proteína, en donde la sola presencia de una variante puede explicar el fenotipo, a veces asociándose a formas predominantemente izquierdas que pueden ser indistinguibles de una miocardiopatía dilatada. A su vez, también existen genes no desmosomales con variantes patogénicas claramente descritas como causa de la enfermedad. Algunos ejemplos son DES, FLNC, PLN y TMEM43.
La miocardiopatía dilatada se define como la presencia la dilatación del ventrículo izquierdo y disfunción sistólica ventricular izquierda en ausencia de condiciones que alteren el llenado, tales como hipertensión, enfermedad valvular o enfermedad arterial coronaria suficiente para causar deterioro de la función sistólica. Se cree que la incidencia de la enfermedad es de 1 en 2.500 individuos de la población general, si bien esta podría ser aún mayor27. Aunque durante muchos años se pensó que la causa era exclusivamente autoinmune o viral, hoy se acepta que más de la mitad de los casos tienen una etiología genética, en los cuales se podría identificar una variante causal o patogénica. Gran parte de estos casos entra en la definición de miocardiopatía dilatada familiar, en la que se requiere que más de un miembro de la familia se encuentre afectado28. La enfermedad presenta gran morbimortalidad asociada, con alta tasa de muerte súbita asociada a arritmias ventriculares (entre 15 y 50% a 5 años)29. El patrón de herencia más frecuente en esta entidad es autosómico dominante, aunque existen formas ligadas al X y recesivas. Hasta la fecha, las variantes de tipo truncamiento en el gen de la Titina, TTN, representan la causa conocida más frecuente de miocardiopatía dilatada30,31. Mutaciones en genes sarcoméricos, filamentos intermedios, proteínas estructurales, factores de transcripción, y proteínas de la membrana nuclear también han sido asociadas al desarrollo de la enfermedad (tabla 2). A medida que aumentan los conocimientos sobre la enfermedad, es mayor la cantidad de genes identificados. Recientemente Ortiz-Genga et al. describieron que las variantes de tipo truncamiento en el gen FLNC asociadas con el desarrollo de un fenotipo solapado de miocardiopatía dilatada/miocardiopatía arritmogénica que se caracteriza por la presencia de arritmias ventriculares frecuentes tienen una alta tasa de muerte súbita en las familias portadoras32.
El consenso de expertos de la Sociedad Europea de Cardiología recomienda el estudio genético en todos los pacientes con defectos de conducción o historia familiar de muerte súbita (recomendación clase I) y en pacientes con miocardiopatía dilatada familiar (recomendación IIa)2,26.
Canalopatías cardiacas
Dentro de esta definición se incluye un grupo heterogéneo de enfermedades primarias que producen algún trastorno de la excitabilidad de las membranas de las células cardiacas. Este grupo de patologías están asociadas a eventos de muerte súbita de causa arrítmica. En su mayoría, son el producto de alteraciones en los genes que codifican para canales iónicos del músculo cardíaco, o proteínas que modulan su función. Aunque comparten al igual que otras cardiopatías heredables una gran heterogeneidad clínica y genética, presentan una alta penetrancia incompleta; solo algunos de los portadores de variantes patogénicas expresan la enfermedad. Este último aspecto dificulta muchas veces los estudios genéticos familiares orientados a confirmar su cosegregación con la enfermedad.
El síndrome de QT largo se caracteriza por un alargamiento del intervalo QT en el electrocardiograma de superficie derivado de una prolongación excesiva de la repolarización ventricular, velocidad de las células miocárdicas para volver a su potencial de membrana en reposo. Esto hace que esté asociado a una alta tasa de muerte súbita por arritmias ventriculares con una incidencia anual, que ronda el 0,9%33. La prevalencia del síndrome de QT largo en población general es aproximadamente 1:2.000 individuos34.
Desde el punto de vista hereditario existen formas autosómicas dominantes, síndrome de Romano-Ward, y recesivas como el síndrome de Jervell y Lange-Nielsen, que incluye sordera neurosensorial congénita y otras alteraciones extracardiacas y solo es producido por mutaciones en los genes KCNQ1 y KCNE1. Variantes patogénicas en al menos 8 genes han sido asociadas con el desarrollo de síndrome de QT largo (tabla 3). El estudio genético puede identificar una variante causal en aproximadamente 75% de los casos, siendo los genes más frecuentemente asociados el KCNQ1, KCNH2 y el SCN5A que corresponden al síndrome de QT largo tipo 1, 2 y 3, respectivamente. Estos tres genes son responsables del 90% de los casos en los pacientes con genotipo positivo26. El consenso de expertos de la Sociedad Europea de Cardiología recomienda al menos el estudio genético para QT largo 1, 2 y 3 en todos los pacientes con diagnóstico probable de QT largo, sin causas que lo puedan explicar (recomendación clase Ic)2,26.
Tabla 3 Genes relacionados con el desarrollo de las diferentes canalopatías y rendimiento del test genético en estas entidades
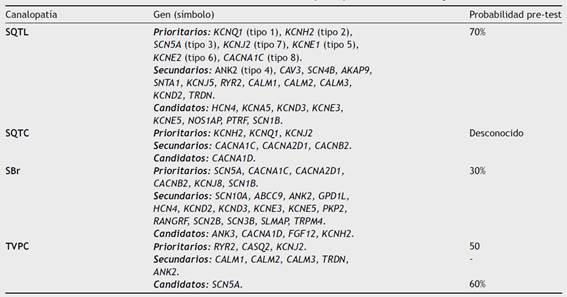
SQTL = síndrome de QT largo; SQTC = síndrome de QT corto; SBr = síndrome de Brugada; TVPC = taquicardia ventricular polimórfica catecolaminérgica.
El síndrome de Brugada es una enfermedad rara caracterizada por anomalías en el electrocardiograma de superficie, elevación con pendiente negativa del ST en precordiales derechas y riesgo elevado de muerte súbita de causa arrítmica. Es responsable del 4 al 12% de los casos de muerte súbita cardiaca y de hasta el 50% de la muerte súbita en corazones estructuralmente normales35.
Se hereda de forma autosómica dominante con penetrancia incompleta y alta heterogeneidad genética. Aproximadamente el 50% de los pacientes con este síndrome tienen historia familiar de la enfermedad. Sin embargo, sólo el 30% son portadores de una variante genética que explique el fenotipo (tabla 3). Las variantes patogénicas en SCN5A, subunidad alfa del canal de sodio, son responsables de aproximadamente el 80% de los casos en los pacientes con genotipo positivo35. El consenso de expertos de la Sociedad Europea de Cardiología recomienda el estudio genético en pacientes con diagnóstico de Brugada tipo 1 (recomendación clase IIa) y lo desaconseja en aquellos con patrón 2 o 3 aislado2,26.
Enfermedades hereditarias de la aorta
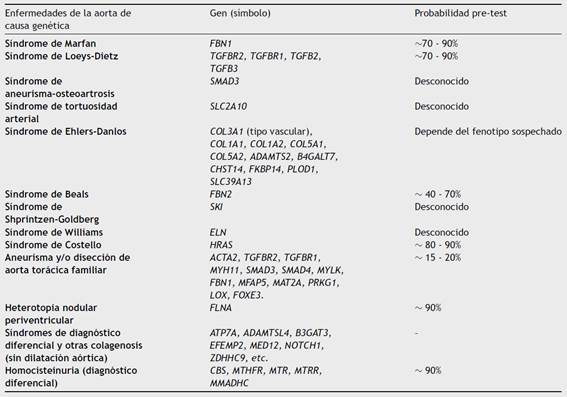
Este grupo de enfermedades incluye algunos trastornos hereditarios del tejido conectivo que pueden llevar a dilatación/aneurisma y/o ruptura/disección de la aorta. Entre estos trastornos se incluyen los síndromes de Marfan, Loeys-Dietz, Ehlers-Danlos tipo vascular, el de aneurisma-osteoartrosis, Shprintzen-Goldberg, el de tortuosidad arterial y el aneurisma o disección de aorta familiar36,37. Estos síndromes pueden presentar características clínicas comunes, como dismorfismo facial, anomalías oculares y alteraciones esqueléticas, que dificultan su diagnóstico clínico. Sin embargo, algunas de estas patologías presentan un comportamiento vascular más agresivo que otras, haciéndose fundamental su adecuado diagnóstico diferencial para realizar una mejor aproximación pronóstica. Los pacientes con síndrome de Loeys-Dietz o de síndrome de aneurisma-osteoartrosis pueden presentar afectación en más de una rama arterial, aorta, coronarias, cerebrales, etc., y eventos de disección incluso sin dilatación vascular previa. La implementación de la secuenciación masiva en muchos casos permite aclarar la etiología genética, orientando, confirmando o replanteando el diagnóstico clínico. En este sentido, se convierte en una herramienta clínica de diagnóstico y de mejor aproximación pronóstica, ya que permite la evaluación y el seguimiento clínico adecuados37,38.
Respecto al rendimiento del test genético en estas enfermedades, utilizando un diseño adecuado de los genes incluidos en el panel a secuenciar y con una orientación clínica apropiada, la confirmación diagnóstica puede llegar a más del 90% en el caso del síndrome de Marfan, 70-95% en el síndrome de Loeys-Dietz y 20% en el síndrome de aneurisma y/o disección de la aorta torácica familiar9. En la actualidad, el estudio genético para los síndromes de Marfan y Loeys-Dietz está incluido dentro de los criterios diagnósticos2,38.
En la tabla 4 se muestran los genes más comúnmente asociados con cada una de estas enfermedades y el rendimiento de la secuenciación masiva en estas entidades.
Conclusiones
Las nuevas tecnologías de secuenciación masiva han revolucionado el estudio de las causas genéticas de la enfermedad en la práctica clínica. La secuenciación masiva permite obtener gran cantidad de información en un periodo corto de tiempo y a un precio razonable, pero requiere la implementación de plataformas bioinformáticas específicas y la conformación de equipos especializados en esta tarea, así como una adecuada interpretación y gestión del conocimiento.
Las cardiopatías heredables son un grupo de enfermedades caracterizadas por una alta heterogeneidad clínica y genética. Todas ellas tienen en común asociarse con riesgo de muerte súbita, muchas veces como única manifestación de la enfermedad.
El estudio genético en cardiopatías heredables permite estudiar la etiología de la enfermedad, dar una mejor aproximación pronóstica, plantear tratamientos más precisos en algunos casos, optimizar el screening familiar, dar consejo genético y permitir a la familia tener una descendencia libre de enfermedad.
La implementación de las nuevas tecnologías de secuenciación junto con una adecuada gestión de la información e interpretación de los resultados constituyen uno de los ejemplos evidentes del nuevo modelo de medicina: predictiva, de precisión y personalizada.

