











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkIntroducción
El síndrome de QT largo representa un grupo de desórdenes electrofisiológicos cardiacos caracterizados por la prolongación del QT y por las anormalidades en la onda T en el electrocardiograma. De acuerdo con la Sociedad Europea de Cardiología, una prolongación del QT es un intervalo mayor de 480 ms tanto en hombres como en mujeres1. El síndrome de QT largo se presenta, en su mayoría, en pacientes jóvenes, y se asocia a síncope, arritmias ventriculares, taquicardia de puntas torcidas y muerte súbita2. A continuación se presentan los casos de dos familias colombianas con varios miembros con síndrome de QT largo, estudio genético completo y presentaciones clínicas diferentes.
Casos
Familia 1
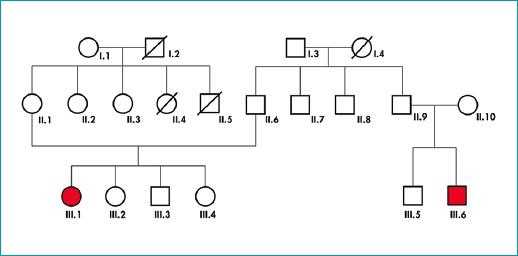
Mujer de 38 años, con episodios de síncope desde la niñez y antecedente familiar de hija y hermana con los mismos síntomas. Se realizan estudios por cardiología, en los que se evidencian Holter de ritmo en sinusal, con extrasístoles ventriculares aisladas. Ecocardiograma TT (transtorácico) con FEVI (fracción de eyección del ventrículo izquierdo) 68%, sin alteraciones estructurales. Mesa basculante positiva para síncope neurocardiogénico, electrocardiograma con QTc prolongado, valorada por Electrofisiología con evidencia de QT largo (569 ms); se descartan causas adquiridas de prolongación de QT. No tomaba ningún medicamento al momento de los estudios. En el análisis genético se identificó variante c.1319dup en heterocigosis en el gen KCNH2, probablemente patogénica, por lo que implantan cardiodesfibrilador (CDI) e inician manejo con betabloqueador (propranolol). Su hija con síncopes desde los 6 años, QTc >500 ms, también heterocigota para la mutación c.1319dup con CDI y manejo con metroprolol succinato. En cuanto a su hermana, no se realiza análisis molecular, presenta episodio de muerte súbita abortada y posterior a este se documenta QTc en 600 ms y requiere implante de dispositivo y tratamiento coadyuvante con propranolol (Fig. 1).

Familia 2
Paciente femenina de 18 años, quien consulta por episodios de presíncope, de características vasovagales de manera repetitiva, no tenía ningún antecedente patológico. El electrocardiograma evidencia QTc en 494 ms, en tanto que el ecocardiograma TT no muestra enfermedad estructural. El análisis genético de análisis genético, en el cual se identifican las variantes ANK2 (c.7636 G>C) y DSP (c.1140+6T>C), por lo que se le implanta monitor de eventos, el cual no ha arrojado ningún resultado patológico hasta el momento. Tiene un primo, quien presentó una arritmia auricular, llevada a ablación; sin embargo, no se ha realizado estudio molecular (Fig. 2).
Discusión
El síndrome de QT largo hereditario se caracteriza por la disfunción de varios canales iónicos que resulta en una repolarización miocárdica anormal con una prolongación del potencial de acción. Las prolongaciones de las duraciones del potencial de acción pueden ser causadas por una disminución de la repolarización y una reducción de la función de los canales de potasio (IKs o IKr) o una persistencia de la entrada de sodio que se extiende a la fase de meseta por un aumento de la función de los canales de Na (INa). Lo anterior predispone a los miocitos a despolarizaciones tempranas o despolarizaciones tardías por la misma prolongación de la duración del potencial de acción3.
La taquiarritmia ventricular que predispone a los eventos cardiacos del síndrome de QT largo es la torsión de puntas, un tipo curioso de taquicardia ventricular que muchas veces es autolimitado y que puede producir síncope, pero que también puede desencadenar en fibrilación ventricular y causar paro cardiaco o muerte súbita.
Se han encontrado hasta 15 subtipos de síndrome de QT largo congénito, cada uno asociado a una mutación en un gen diferente. Más de un 85% de los pacientes con síndrome de QT largo se encuentran entre los subtipos 1, 2 y 3. En su mayoría tienen un patrón de herencia autosómico dominante, también conocido como síndrome Romano-Ward4.
La prolongación del QT puede ser causada por muchas condiciones, entre las que se incluyen alteraciones hidroelectrolíticas, medicamentos, cardiopatía estructural, miocarditis, hipertrofia ventricular y anormalidades del QRS, por lo que una evaluación detallada para excluir estas causas debe realizarse para considerar el diagnóstico de síndrome de QT largo5,6.
Uno de los siguientes tres criterios se requieren para realizar el diagnóstico de síndrome de QT largo.
Un puntaje de Schwartz (Tabla 1) de al menos 3.5 en ausencia de causas secundarias de prolongación del QT.
Un QTc entre 480 - 500 ms en un ECG de 12 derivaciones en ausencia de causas secundarias de prolongación del QT.
Presencia de variante genética patogénica para síndrome de QT largo independiente del QTc. Este estudio se puede solicitar utilizando múltiples paneles genéticos disponibles en nuestro país, con el código CUPS 908412 equivalente al diagnóstico molecular de enfermedades, se pueden solicitar estos paneles.
Tabla 1 Descripción molecular de los casos afectados
| Gen | Variación | Significado | Muerte súbita/síncope | CDI | ||
|---|---|---|---|---|---|---|
| Familia 1 | Caso 1 | KCNH2 | c.1319dup | Variante patogénica | Si | Si |
| Caso 2 | KCNH2 | c.1319dup | Variante patogénica | Si | Si | |
| Familia 2 | Caso 1 | ANK2 DSP |
c.7636G>C c.1140+6T>C |
Significado desconocido/probablemente benigna | No | No |
Para realizar el análisis molecular del caso índice, en Colombia, podemos contar con la secuenciación del exoma/genoma7, secuenciación de un panel de genes o un gen individual, el exoma trío, o PCR (Reacción en cadena de la polimerasa) y secuencia del fragmento donde se encuentra la mutación8. Como las enfermedades cardiovasculares no son enfermedades clásicas mendelianas, y varias son poligénicas, es difícil tomar una decisión sobre cual gen estudiar, por lo no se recomienda la realización de secuenciación de un solo gen8, los paneles de genes o la secuenciación del exoma son opciones válidas, útiles9. Si ya se tiene el diagnóstico molecular en el caso índice, se puede utilizar la PCR de la mutación en los familiares de primer grado o con sospecha de estar afectados.
Es importante la adecuada interpretación, ya que cada prueba conlleva un grado de incertidumbre sobre si una condición se desarrollará, cuándo se desarrollará y qué tan grave será10.
En cuanto al tratamiento, los Betabloqueadores son la terapia de primera línea en pacientes con síndrome de QT largo sintomáticos11. El agente más estudiado y efectivo es el propanolol (dosis de 2 a 3 mg/kg/día) y el Metoprolol es definitivamente el menos efectivo por lo que no se recomienda para el tratamiento de síndrome de QT largo12. El nadolol también se utiliza con frecuencia por tener una vida media más larga (dosis 1 a 1.5 mg/kg día) y podría incluso llegar a ser más efectivo que el propanolol13. La otra terapia importante para el Síndrome de QT Largo incluye el implante de un cardiodesfibrilador (CDI) en pacientes con antecedente de muerte súbita, con persistencia de sincope en terapia médica o con una mutación de alto riesgo14. La denervación simpática cardiaca izquierda (DSCI) es una opción efectiva y simple pero poco utilizada en el tratamiento de arritmias cardiacas hereditarias, esta intervención impacta en la calidad de vida de los pacientes con síndrome de QT largo sin embargo no impacta en la mortalidad como si lo hace el implante de CDI15.
A la familia del primer caso se le realizó secuenciación de nueva generación, para regiones codificantes y los sitios de splicing de 12 genes conocidos asociados a síndrome de QT largo: KCNQ1, KCNH2, SCN5A, ANK2, KCNE1, KCNE2, KCNJ2, CACNA1C, CAV3, SCN4B, AKAP9, SNTA1. Siendo positiva para la variante c.1319dupC en heterocigosis en el gen KCNH2. Este gen (KCNH2), localizado en el cromosoma 7q6.1, codifica la subunidad a del canal de potasio IKr; la disfunción de este canal disminuye la corriente saliente de potasio durante la fase 3 del potencial de acción, prolongando su duración16. Se asocia a síndrome de QT largo tipo 2, con características de mal pronóstico como síncope y muerte súbita17,18.
En la base de datos HGMD (The Human Gene Mutation Database http://www.hgmd.cf.ac.uk/)19 y en Broad Institute ExAC database http://exac.broadinstitute.org20, esta mutación no ha sido reportada, sin embargo, en ClinVar cumple condiciones de ser una variante patogénica21,22. Esta variante produce un codón de parada prematuro, dando origen a una proteína KCNH2 truncada en aminoácido 441 (p.Pro441Alafs*78), por lo que es muy probablemente patogénica, debido a que este aminoácido se encuentra en el bucle extracelular del receptor entre los dominios S1 y S2, con pérdida de múltiples regiones posterior a la mutación.
Mutaciones similares se han reportado en una posición similar, como el c.1280A>C que causa una proteína truncada en el aminoácido p.Y427S*, debido a un codón de parada, este hallazgo, en un paciente heterocigótico, corresponde a un fenotipo dominante de muerte súbita cardiaca de un síndrome de QT largo tipo 223.
En la posición c. 1319 C>T, p.P440L*, una mutación missense, desencadenó en un paciente un Síndrome de QT largo24. Una mutación en la posición del aminoácido E444D del gen KCNH2, en una familia de China, desarrolló Síndrome de QT largo tipo 2, caracterizado por QTc > 500 ms, síncope y torsade de puntas25. Por lo anterior es posible afirmar que las alteraciones cerca al aminoácido 441 presentan desenlaces clínicos de mal pronóstico. Y, si el paciente tiene más de una mutación en el KCNH2 es posible que empeore el pronóstico26. En un estudio publicado por Mullally et al. en el 2013, se documenta que los pacientes con más de una mutación, presentaban un QTc más largo, y presentaron más posibilidad de desarrollar eventos cardiacos potencialmente mortales 23% vs. 11%, con una p 0.03127.
En el segundo caso, se realiza un exoma dirigido con objeto de identificar las variantes en 74 genes, identificando variantes en el gen ANK2 c.7636 G>C (p.Val2546Leu) y DSP (c.1140+6T>C splicing).
El gen ANK2, localizado en el cromosoma 4q25-q26, codifica una de las proteínas de la familia de la anquirina, que tiene como función unir las proteínas integrales de la membrana al citoesqueleto subyacente de la espectrina y la actina, participa en funciones como: motilidad celular, activación, proliferación y mantenimiento de la membrana celular28. Hay 127 mutaciones tipo missense/nonsense descritas en la base de datos HGMD, la mayoría asociadas a Síndrome de QT largo tipo 4, la variante identificada en la paciente de la familia 2 c.7636 G>C (p.Val2546Leu), no ha sido reportada previamente19,20. Esta variante, que altera la secuencia de aminoácidos, no modifica la longitud del péptido, las características, ni la función de la proteína, se trata de una variante de cambio de sentido, sin asociación a un fenotipo específico, por lo que, in silico es catalogada como benigna29.
En ClinVar encontramos una variante en la posición c.7620C>A, la cual también es informada como benigna, sin datos clínicos30, Así mismo la variante c.7650T>G (p.Val2550=) que se ubica corriente debajo de la posición 7636 no muestra asociación fenotípica y es probablemente benigna31.
El gen DSP, localizado en el cromosoma 6p24.3, codifica para la proteína desmoplaquina componente crítico del desmosoma del musculo cardiaco, que funciona para mantener la integridad estructural con las células adyacentes, se han descrito 11.129 variantes en ClinVar, principalmente relacionadas con displasias ectodérmicas, epidermólisis, y en el sistema cardiovascular asociadas a cardiopatía dilatada y displasia arritmogénica de ventrículo derecho32, la variante de splicing identificada en heterocigosis en la muestra ha sido previamente documentada en la base de datos ClinVar presentando conflicto de interpretación entre variante probablemente benigna y variante de significado incierto21, al buscarla en otra base de datos, como PolyPhen se cumple con criterios de variante benigna29. Se observa que esta paciente ha presentado presíncopes, y no ha tenido eventos de síncope, arritmias ventriculares o muerte súbita. Por lo que, en este caso, el análisis molecular no explica los síntomas, ni la prolongación del QT. Se podría ampliar el número de genes para secuenciación o buscar un efecto aditivo entre las dos variantes que me prolonguen el QTc, sin causar eventos cardiovasculares mayores.
Podemos ver con estos casos, las diferencias en cuanto a la presentación clínica de esta enfermedad (síncope, presíncope o muerte súbita) y la importancia del diagnóstico genético para el pronóstico. Encontrando que una mutación en el gen KCNH2 tiene peor pronóstico17,27. Además, dos pacientes portadores de mutaciones solo presentaban presíncope y uno con test de mesa basculante positivo, haciendo énfasis en que así no se encuentre entre los criterios diagnósticos debemos revisar cuidadosamente el QT en los pacientes que manifiesten este síntoma, tanto en el electrocardiograma, en el estudio de Holter y en la mesa basculante. En un estudio de 68 pacientes con síndrome de QT largo, se realizó medición del QT antes y después de la bipedestación, y se documentó durante la taquicardia máxima, el intervalo QT corregido aumentó en 50 +/- 30 ms en el grupo de control y en 89 +/- 47 ms en el grupo LQTS (p <0,001). Las curvas características del receptor mostraron que la prueba agrega valor de diagnóstico. Por lo que se comprueba que los pacientes tienen mala adaptación a los cambios bruscos de frecuencia cardiaca33.
Los corazones de todos los casos índice eran estructuralmente sanos en el Ecocardiograma TT y la terapia con CDI y Betabloqueadores, los cuales actúan disminuyendo el intervalo QT, han mejorado el pronóstico y la sobrevida de todos.
El tratamiento y seguimiento de estas familias, debe ser llevado por un cardiólogo especialista en canalopatías y miocardiopatías hereditarias, junto con la ayuda de un genetista, dada la penetrancia incompleta, expresividad variable y la difícil interpretación de los resultados34. La asesoría genética es importante para la adecuada obtención de antecedentes familiares, la correcta elaboración de un árbol genealógico, entender la enfermedad genética, afrontarla, comprender el modo de herencia, consecuencias en la descendencia y tomar medidas al respecto35.
Tabla 2 Puntuación de Schwartz
| Puntos | |
|---|---|
| Hallazgos electrocardiográficos | |
| QTc (ms) reposo | |
| > 480 ms | 3 |
| 460-479 ms | 2 |
| 450 - 459 ms (hombres) | 1 |
| QTc a los 4 minutos de recuperación en prueba de esfuerzo | 1 |
| Torsión de puntas | 2 |
| Alternancia de onda T | 1 |
| Onda T mellada en 3 derivaciones | 1 |
| Frecuencia cardiaca baja para la edad | 0.5 |
| Historia clínica | |
| Síncope | |
| Con estrés | 2 |
| Sin estrés | 1 |
| Sordera congénita | 0.5 |
| Historia familiar | |
| Familiar con diagnóstico definitivo de síndrome de QT largo | 1 |
| Muerte súbita inexplicada en un familiar de primer grado < 30 años | 0.5 |
< 1 punto baja probabilidad de síndrome de QT largo. 1-3 puntos probabilidad intermedia para síndrome de QT largo. ≥ 3.5 alta probabilidad de síndrome de QT largo. Adaptado de Ku CS, et al.6
Fortalezas y limitaciones del estudio
En este artículo queremos resaltar la importancia de la elección del análisis molecular que vamos a utilizar, de la adecuada interpretación de las pruebas genéticas y todas las herramientas en las que nos podemos apoyar para el tratamiento y la asesoría de los pacientes, debido a que en ocasiones esto es desconocido para los cardiólogos.
Como limitaciones es claro que tenemos una muestra pequeña y que el seguimiento de los pacientes no ha sido a largo plazo.
Conclusión
El síndrome de QT largo congénito es una patología compleja, sus modos de presentación son potencialmente fatales y el proceso diagnóstico completo es largo y en nuestro país difícil de realizar y de interpretar. Es importante conocer cuando es necesario el estudio genético y los diferentes métodos diagnósticos aptos para cada caso, ya que es diferente cuando el paciente es el primer caso en la familia o cuando ya se cuenta con un familiar que tiene una mutación identificada y éste es asintomático, como lo vimos en los casos presentados. Es por esto, que la asesoría genética juega un papel muy importante en el acompañamiento de estos casos, debido a que analizan e identifican las variantes genéticas que afectan la función de las proteínas y su intervención tiene un impacto directo en el tratamiento de los pacientes y sus familiares.

