











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkCaso 1
Mujer de 30 años, con cuadro de dos años de evolución, consistente en deterioro de la clase funcional hasta II/IV de acuerdo con la clasificación de la NYHA; edema en miembros inferiores, disnea paroxística nocturna y ortopnea, quien había consultado en múltiples ocasiones y había sido hospitalizada varias veces por dicha sintomatología, por lo cual era conocida en la institución donde consultó por primera vez por cuadro de dos meses consistente en agudización de los síntomas y deterioro de clase funcional hasta IV/IV de la NYHA. Ingresó a la unidad de cuidados intensivos por falla cardiaca, con perfil congestivo y de hipoperfusión, requerimiento de soporte inotrópico e inicio de terapia de reemplazo renal con hemodiálisis por síndrome cardiorrenal asociado.
Como antecedentes se diagnosticó síndrome de ALCAPA ocho meses antes de su ingreso e insuficiencia renal crónica estadio 5 (glomeruloesclerosis focal y segmentaria por resultado de biopsia del 16 de marzo de 2015), cardiopatía isquémica en fase dilatada con fracción de eyección del ventrículo izquierdo (FEVI) del 10 %, portadora de cardiodesfibrilador (CDI) como prevención primaria de muerte súbita desde abril de 2015, y en manejo con antagonistas de los receptores de angiotensina II, betabloqueador cardioselectivo, antagonistas de aldosterona y diurético de ASA.
Luego de compensar su estado clínico fue llevada a estudios de extensión para evaluar su condición cardiovascular. Se realizó nuevo ecocardiograma transtorácico que informó cardiopatía dilatada con hipoquinesia severa y compromiso severo de la función ventricular, fracción de eyección del 10 %, disfunción diastólica de tipo restrictivo, dilatación auricular izquierda severa, dilatación ventricular derecha severa con disfunción sistólica moderada a severa, insuficiencia tricúspide moderada, PSAP 55 mm Hg, dilatación severa de la arteria pulmonar, insuficiencia pulmonar moderada, electrodo con CDI en cavidades derechas.
Se realizó arteriografía coronaria izquierda, la cual mostró nacimiento anómalo de la arteria pulmonar en el segmento proximal. Al realizar inyección en la arteria coronaria derecha se apreció abundante circulación colateral que llenó todo el árbol coronario izquierdo, incluyendo el tronco, y se evidenció el nacimiento anómalo anotado, sin lesiones en su luz. Coronaria derecha dominante, dilatada, con nacimiento normal, sin lesiones obstructivas. Cateterismo derecho: aurícula derecha 25 mm Hg; ventrículo derecho 70/20 mm Hg; arteria pulmonar 71/20(39) mm Hg y capilar pulmonar 30 mm Hg. Se complementaron imágenes de angiografía con angio-TAC de aorta torácica y de arterias coronarias para evaluar la posibilidad de tratamiento quirúrgico, la cual mostró síndrome de ALCAPA (Fig. 1), con origen de la arteria coronaria izquierda desde el margen inferior del tercio medio del tronco principal de la arteria pulmonar; cardiomegalia global; derrame pericárdico y fracción de eyección del ventrículo izquierdo del 24.1 %. Se evaluó viabilidad para evaluar beneficio de una intervención quirúrgica a través de perfusión miocárdica con estrés farmacológico, la cual reportó necrosis en territorio de la arteria descendente anterior, en extensión aproximada del 25 % de la masa miocárdica, sin dilatación transitoria.
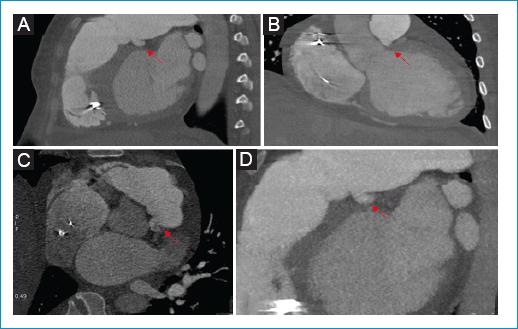
Figura 1 A: reconstrucción sagital oblicua de Angiotac gatillado que demuestra origen anómalo de la arteria coronaria izquierda desde el tronco principal de la arteria pulmonar (flecha roja). B: reconstrucción coronal de Angiotac demostrando origen anómalo desde la pared inferior del tronco principal de la arteria pulmonar (flecha roja). C: corte axial de angiotac gatillado demostrando origen anormal de la arteria coronaria principal izquierda y aumento del diámetro del tronco principal de la arteria pulmonar. D: reconstrucción sagital MIP demostrando origen anómalo de arteria coronaria principal (flecha roja) desde la pared inferior del tronco principal de la arteria pulmonar.
Con estos hallazgos se consideró que no se beneficiaba de la intervención quirúrgica de la anomalía coronaria, así que su tratamiento se encaminó a optimizar el manejo farmacológico y a evaluar la posibilidad de un trasplante dual corazón-riñón.
Caso 2
Paciente de 61 años, con cuadro de dolor torácico irradiado a cuello y maxilar, aproximadamente de 5 años de evolución, de carácter progresivo, con antecedentes de diabetes mellitus tipo 2, hipertensión arterial, dislipidemia e hipotiroidismo en tratamiento con losartán, ácido acetil-salicílico, atorvastatina y levotiroxina. Se realizaron estudios con perfusión miocárdica, los cuales mostraron fracción de eyección del 41 %, VFD 170 y VFS 100, positiva para isquemia anterior con extensión del 6 %. El ecocardiograma mostró cardiopatía dilatada, compromiso moderado de la función sistólica del ventrículo izquierdo del 40 %, disfunción diastólica tipo I, esclerosis valvular mitral y aórtica con insuficiencia mitral leve, y crecimiento auricular izquierdo. Se llevó a arteriografía coronaria en la que se evidenció nacimiento anómalo del tronco coronario izquierdo el cual emergía de la arteria pulmonar; además, arteria coronaria derecha severamente dilatada, con abundante llenado a la arteria coronaria izquierda, dilatación moderada del ventrículo izquierdo e hipertensión pulmonar. Posteriormente, se hizo angio-TAC de aorta torácica y de arterias coronarias (Fig. 2), en la que se confirmó anomalía de origen de la arteria coronaria izquierda, síndrome de ALCAPA (arteria coronaria izquierda: origen desde el tronco principal de la arteria pulmonar con un tronco común de aproximadamente 5 mm de longitud con diámetro de 6.5 mm), circulación coronaria derecha dominante, importante tortuosidad, ectasia de la arteria coronaria derecha y fracción de eyección del ventrículo izquierdo del 58.3 %). Se documentó fibrilación auricular paroxística, por lo cual fue evaluado por cirugía cardiovascular, quienes realizaron injertos aorto-coronarios N2 de safena a la arteria descendente anterior y de safena a la obtusa marginal en "T", además de aislamiento de venas pulmonares más ligadura de la aurícula izquierda con atriclip off pump. El paciente tuvo una evolución adecuada posterior a la intervención. Se hizo seguimiento en controles por Cardiología hasta completar un año posterior a la intervención, cuya evolución fue satisfactoria y resultó asintomático desde el punto de vista cardiovascular.

Figura 2 A: reconstrucción multiplanar coronal de Angiotac gatillado que muestra origen anómalo de la arteria coronaria principal izquierda a partir del tronco principal de la arteria pulmonar (flecha roja). B: corte axial de Angiotac que muestra origen anómalo de la arteria coronaria principal izquierda a partir de la arteria pulmonar principal (flecha roja). C: reconstrucción volumétrica que demuestra arteria coronaria tortuosa y ectasia con origen en el tronco principal de la arteria pulmonar (flecha blanca).
Discusión
El síndrome de ALCAPA, llamado así por sus siglas en inglés (Anomalous origin of the Left Coronary Artery from the Pulmonary Artery), es una rara cardiopatía congénita de la cual se entenderán dos tipos: el que se presenta en la infancia y el de la edad adulta1.
En la infancia es una de las causas más comunes de falla cardiaca, isquemia e infarto de miocardio2. Su incidencia es de 1 por 300.000 nacidos vivos. La mortalidad en pacientes no tratados es del 35 al 85 % en el primer año de vida3.
Ocasionalmente, los pacientes pueden sobrevivir hasta la edad adulta, cuando no han recibido tratamiento en la infancia; puede manifestarse como infarto de miocardio, disfunción ventricular izquierda e insuficiencia mitral o isquemia silente que podría llevar a muerte súbita2.
Durante la etapa fetal y neonatal temprana, la anomalía es bien tolerada debido a que la presión pulmonar es igual a la sistémica. Posteriormente, en la adaptación de la vida intrauterina a la extrauterina, la presión arterial pulmonar disminuye, al igual que el flujo en la arteria coronaria izquierda (ACI), por lo que se presenta isquemia e infarto de miocardio. En el momento de la disminución del flujo de la ACI se desarrolla circulación colateral de la arteria coronaria derecha a la izquierda. Cuando está bien establecida se desarrolla el síndrome de ALCAPA del tipo de la edad adulta3; sin embargo, el aporte al ventrículo izquierdo es insuficiente, en especial en la región subendocárdica, por lo que se produce isquemia crónica con disfunción ventricular, pudiendo estar inicialmente asintomático2.
En el abordaje diagnóstico se han utilizado diferentes técnicas para su detección, algunas invasivas, como la arteriografía coronaria4, y otras no invasivas, como la TAC de arterias coronarias5,6, considerada esta última como de primera línea para el diagnóstico y seguimiento, ya que muestra de manera adecuada la anomalía y tiene el beneficio de aportar información adicional en caso de requerirse manejo quirúrgico2. Como parte de los estudios de extensión se han utilizado la RMN de corazón2 y la perfusión miocárdica5 para determinar la viabilidad miocárdica y así determinar si los pacientes pueden beneficiarse del tratamiento quirúrgico.
Los hallazgos imagenológicos primarios consisten en la visualización directa de la ACI saliendo de la cara antero-lateral izquierda de la arteria pulmonar; posteriormente, sigue el curso hacia el surco interventricular y se ramifica, formando las arterias descendente anterior y circunfleja; esta es la distribución usual. También puede observarse flujo retrógrado de la ACI a la arteria pulmonar en las imágenes de RMN de corazón, y jet de regurgitación por medio de SSFP cine (secuencia eco de gradiente en modo cine)2. Como hallazgos imagenológicos secundarios figuran: dilatación y tortuosidad de las arterias coronarias derecha e izquierda, evidencia de ramas colaterales dilatadas provenientes de la ACD (arteria coronaria derecha) a la ACI, hipertrofia, dilatación y alteraciones de la contractilidad ventricular izquierda, degeneración mixomatosa y disfunción isquémica de los músculos papilares y adyacentes, la cual ocasiona insuficiencia y prolapso de la válvula mitral.
El realce tardío en RMN por isquemia subendocárdica crónica, hallazgo importante en pacientes asintomáticos o con diagnóstico incidental de la enfermedad, es un factor predictor de arritmias malignas, y en el seguimiento, si se observa empeoramiento de esta condición, debe evaluarse la posibilidad de tratamiento quirúrgico2.
La corrección quirúrgica es considerada el tratamiento estándar, a menos que exista alguna contraindicación6. El método de elección es el reparo quirúrgico del defecto realizando redireccionamiento con parche o traslocación de la arteria coronaria izquierda anastomosándola en la aorta, con muy buenos resultados a largo plazo6, la cirugía de revascularización queda reservada para casos en que estas técnicas no son posibles y debe asociarse a ligadura del tronco de la coronaria. Otras opciones quirúrgicas incluyen trasplante cardiaco y cierre percutáneo de ACI anómala2,6.

