











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
La hipertrigliceridemia (HTG) es un problema que se presenta con frecuencia en la práctica clínica. Su prevalencia en los adultos es cercana al 10%. La prevalencia de HTG leve a moderada es paralela a la prevalencia de la obesidad y la diabetes mellitus tipo 2; su aumento, por lo tanto, no es sorprendente en las últimas décadas1,2. La gravedad de la HTG varía ampliamente y hasta la fecha no se ha establecido una clasificación uniforme de la afección1-3. La mayoría de las personas afectadas (80-90%) tienen valores de triglicéridos (TG) moderadamente aumentados, es decir, entre 1.7 mmol/l (150 mg/dl) y 4.6 mmol/l (400 mg/dl)2. La HTG grave, definida como la concentración de TG en plasma > 10 mmol/l (> 885 mg/dl) es menos común, con una prevalencia que varía del 0.10% al 0.20%, mientras que la HTG muy grave, definida como TG > 20 mmol/l (> 1770 mg/dl), es aún más rara (prevalencia del 0.014%)1. Al interpretar las concentraciones de TG se debe tener en cuenta que estos valores umbral se aplican a los TG en ayunas2.
Como ya se dijo, la HTG está estrechamente asociada con la presencia de obesidad, síndrome metabólico y diabetes mellitus. Por ejemplo, hasta el 50% de los pacientes con diabetes tipo 2 tienen HTG concomitante. Independientemente de esto, con frecuencia existe una predisposición genética que conduce, en combinación con factores de estilo de vida, a HTG. Esta predisposición suele ser poligénica. El espectro varía desde una disposición que resulta en HTG solo en presencia de sobrepeso considerable o de consumo excesivo de alcohol hasta mutaciones graves y muy raras. Estas mutaciones se pueden encontrar en genes que codifican proteínas tales como la lipoproteína lipasa (LPL) y las apolipoproteínas (Apo) A-V, CII y CIII, las cuales pueden conducir a HTG sumamente grave en la infancia, incluso en ausencia de factores adicionales, como el síndrome de quilomicronemia familiar (FCS, familial chylomicronemia syndrome)2,4.
El FCS también se conoce como hiperlipoproteinemia tipo I o deficiencia de LPL5-7. Esta es una enfermedad autosómica recesiva rara (una o dos personas por cada millón) del metabolismo del quilomicrón asociada con una importante elevación de la concentración de TG circulantes, así como una mayor incidencia de pancreatitis, pancreatitis recurrente y mortalidad asociada con pancreatitis aguda3.
Desafortunadamente, esta enfermedad a menudo es desconocida por muchos proveedores de atención médica, lo que lleva a retrasos en el diagnóstico y tratamientos inadecuados5-7. Por lo tanto, el objetivo de la presente revisión es ampliar la comprensión de estas afecciones, proporcionando una visión general basada en la literatura.
Obtención de los datos
Se identificaron todos los artículos relevantes en PubMed y Google Scholar usando términos de búsqueda tales como "severe hypertriglyceridaemia", "familial hyperchylomicronemia", "familial chylomicronemia síndrome", "lipoprotein lipase deficiency", "type I hiperlipoproteinemia", "type I hiperlipidemia", "chylomicronemia" y términos relacionados que no se refieren directamente al FCS, incluyendo "HTG induced acute pancreatitis". También se revisaron las listas de referencias de cada publicación seleccionada, así como la literatura disponible. En este trabajo nos centramos en publicaciones de los últimos 5 años (2015-2020). Es de importancia mencionar que la disponibilidad de literatura en español es muy limitada.
Metabolismo normal de los triglicéridos
Las lipoproteínas ricas en TG, es decir, quilomicrones y lipoproteínas de muy baja densidad (VLDL), son complejos esféricos compuestos de lípidos centrales (TG y ésteres de colesterol) con apolipoproteínas de superficie, fosfolípidos y colesterol libre. Los TG exógenos (dietéticos) se transportan en quilomicrones derivados del intestino, mientras que los TG de origen endógeno circulan en las VLDL derivado hepáticamente (Fig. 1)1,8,9. En cualquier nivel de secreción de quilomicrones y VLDL, la eficacia de la lipólisis mediada por LPL y la captación hepática de partículas remanentes determinará la cantidad de TG circulantes en ayunas y posprandial. La lipólisis mediada por lipasa de lipoproteínas ricas en TG recientemente secretadas es un proceso saturable, y a medida que aumentan las tasas de secreción y las cifras de TG en plasma disminuye la lipólisis1,10. En general, el aumento de la producción de VLDL es el factor iniciador más común para la HTG1.
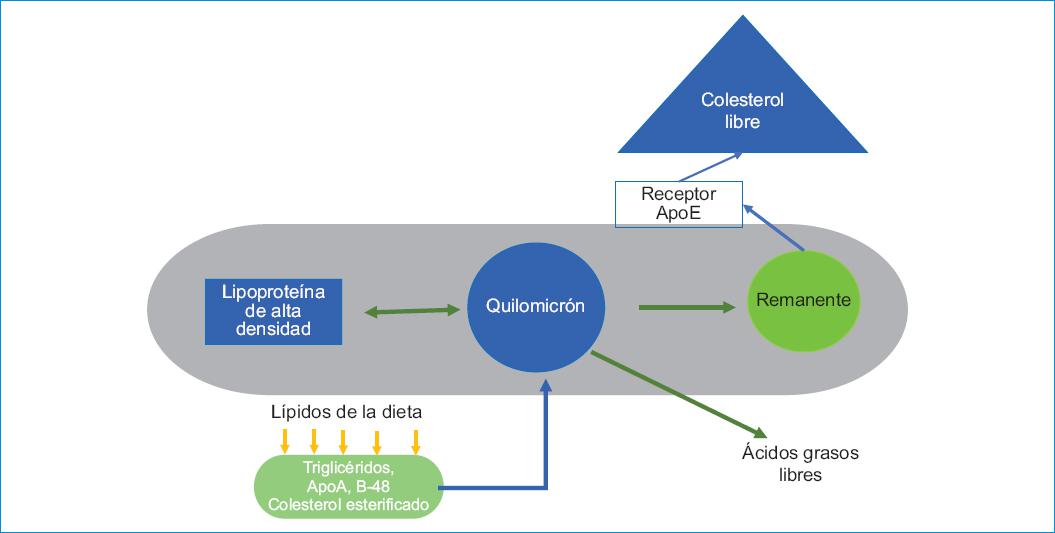
La LPL es una enzima que elimina los TG de las lipoproteínas ricas en TG y los descompone en ácidos grasos libres para usarlos como energía. Se produce en los tejidos adiposos y musculares, y se secreta en el espacio intersticial de estos tejidos. Después de que se sintetiza LPL, para ganar más capacidad funcional necesita cofactores y proteínas de transporte. El factor de maduración de la lipasa 1 (LMF-1) facilita la secreción de LPL del retículo endoplásmico de adipocitos y miocitos en forma activa como homodímero. Después de eso, se requiere de la proteína 1 de unión a lipoproteína de alta densidad anclada (GPIHBP1) para transferir LPL desde el espacio intersticial a través del endotelio capilar y anclarlo a la superficie capilar. Para volverse completamente funcional, la LPL se une a la Apo C-II. Adicionalmente, la Apo A-V también mejora la actividad de la LPL al actuar como cofactor para la interacción de la LPL y la Apo C-II. Por otra parte, cuando la Apo C-III está presente en lipoproteínas ricas en TG, actúa para inhibir la función de la LPL y también tiene otras funciones, incluida la inhibición de la proteína triglicérido lipasa hepática. Las mutaciones en genes que codifican proteínas que causan fallas en la producción de LPL (como LMF-1), el transporte de LPL (como GBIHBP1) o la activación de la LPL (como Apo C-II y Apo A-V) pueden afectar la función de la LPL y el exceso de quilomicrones en la circulación4,7,10.
Quilomicronemia
La quilomicronemia es la acumulación en el torrente sanguíneo de quilomicrones, que como ya se mencionó son partículas de lipoproteínas producidas por los enterocitos después de una comida. En condiciones fisiológicas, los quilomicrones se eliminan rápidamente del plasma mediante la acción de la LPL. Los quilomicrones se convierten en quilomicrones remanentes, que a su vez son absorbidos por receptores celulares específicos11.
La quilomicronemia en ayunas se caracteriza por unos valores de TG en plasma > 10 mmol/l (> 885 mg/dl), clasificado como HTG grave por el Panel de Consenso de la Sociedad Europea de Aterosclerosis. La quilomicronemia es una causa poco reconocida de HTG grave, por lo cual se decidió abordarla en esta revisión4.
El síndrome se puede clasificar en dos grupos principales4,10,12,13: FCS y síndrome de quilomicronemia multifactorial (MCS).
El FCS, anteriormente conocido como hiperlipoproteinemia de tipo 1, actualmente se cree que es un trastorno monogénico1,5. Genera HTG constante, con descompensaciones recurrentes en ausencia de evidencia de factores secundarios. Además de la deficiencia homocigótica de LPL, deben considerarse las mutaciones homocigotas de pérdida de función en los genes APOC2, APOA5, GPIHBP1, LMF1 y G3PDH1, así como mutaciones heterocigotas compuestas que involucran LPL y estos genes (Fig. 2)9,11. Se hereda de forma autosómica recesiva, y frecuentemente se manifiesta en niños o adolescentes. La enfermedad se asocia con falta de crecimiento, pancreatitis, TG altos y valores bajos de otras fracciones lipídicas. Además, suele ser refractaria a la terapia de reducción de TG, lo que dificulta el tratamiento4.
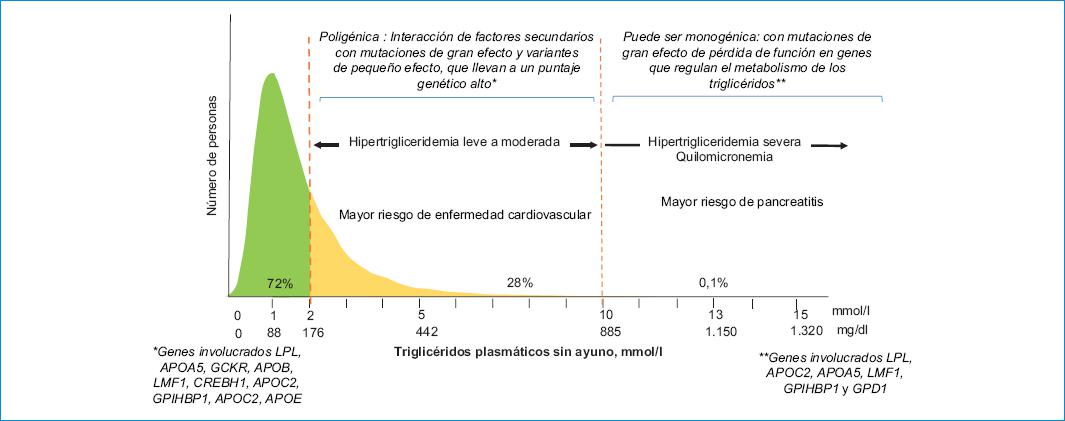
Por el contrario, el MCS, antes conocido como hiperlipoproteinemia de tipo 5, es un trastorno mucho más frecuente, que incluye una variedad de factores patológicos combinados genéticos y ambientales, como son la diabetes tipo 2, el abuso de alcohol y el uso de estrógenos, entre otros4,10,12,13. Los pacientes pueden tener una combinación de una mutación heterocigota de pérdida de función o variantes frecuentes patógenas probables en genes que aumentan los TG, produciendo HTG grave (oligogénica o poligénica). Sin embargo, en tales casos, la hiperquilomicronemia a menudo es transitoria y la baja actividad de LPL es inconstante. Entre las descompensaciones recurrentes, estos pacientes presentan principalmente HTG leve. Este fenotipo se observa a menudo en mutaciones heterocigotas de pérdida de función de APOA55. La quilomicronemia poligénica generalmente se manifiesta más tarde en la vida y suele responder a las terapias disponibles para reducir los TG (Fig. 2)4.
Manifestaciones clínicas
El fenotipo aparentemente similar entre MCS y FCS significa que se puede pasar por alto o no diagnosticar esta última, convirtiéndose en una causa poco conocida y poco diagnosticada de HTG grave5.
Las manifestaciones asociadas con el FCS son bastante heterogéneas e inespecíficas. Los hallazgos clásicos y más frecuentes incluyen xantomas eruptivos, lipemia retiniana, hepatoesplenomegalia o esplenomegalia, náuseas, vómitos, dolor abdominal recurrente y falta de crecimiento2,5,7. Además, se han documentado síntomas neurológicos, como confusión transitoria, irritabilidad, problemas de memoria, demencia y depresión5,7. Los pacientes tienden a tener un peso corporal más bajo debido a la restricción de su ingesta de alimentos debido al dolor abdominal4, y a menudo muestran un relativo agotamiento del tejido adiposo sin evidencia de obesidad central7. El impacto de la enfermedad es tal que la calidad de vida y las actividades diarias, incluido el trabajo, pueden verse negativamente afectadas5,7.
Los xantomas eruptivos, en general presentes en las nalgas, los hombros y las superficies extensoras de las extremidades, representan una respuesta inflamatoria a la acumulación de lípidos en los tejidos y se observan en aproximadamente un tercio de los pacientes. En la lipemia retiniana, los vasos retinianos aparecen blanqueados en la fundoscopia, pero la visión no se ve afectada. La hepatoesplenomegalia se debe a la infiltración de macrófagos en respuesta al depósito de quilomicrones5.
Los pacientes con FCS tienen un alto riesgo de episodios de pancreatitis aguda, potencialmente mortales y que pueden ser recurrentes si los TG no se controlan de manera óptima5. Los episodios recurrentes de pancreatitis aguda pueden conducir a pancreatitis crónica, así como a insuficiencia pancreática exocrina y endocrina. Aunque el mecanismo preciso por el cual la HTG o la quilomicronemia conducen a pancreatitis aguda es desconocido, los mecanismos propuestos incluyen la formación excesiva de radicales oxidados a partir de ácidos grasos libres catabolizados aberrantemente por la lipasa pancreática o la inflamación provocada por la sobrecarga mitocondrial, así como la hiperviscosidad de la sangre por los quilomicrones circulantes excesivos que llevan a isquemia local y acidosis dentro del páncreas2,5,7.
Diagnóstico
No hay unos criterios de diagnóstico unificados para el FCS5,14. Existen varios algoritmos propuestos, de los que algunos recomiendan el uso de secuenciación genética diagnóstica, otros proponen un puntaje fenotípico de FCS y otros sugieren diagnosticar la condición en función de las características clínicas después de la exclusión de la HTG secundaria o poligénica14,15.
Antes de que las pruebas genéticas estuvieran disponibles, una prueba tradicional para diagnosticar la deficiencia de LPL era la medición de la actividad de LPL en plasma después de la inyección de heparina5,7. Una marcada reducción o la ausencia de actividad de LPL (es decir, ≤ 20% de lo normal) a menudo se usaba en el pasado para documentar la deficiencia de LPL. Sin embargo, este ensayo ya no suele estar disponible en el entorno clínico y es relativamente poco confiable, ya que el rendimiento varía en gran medida dependiendo de la experiencia del laboratorio de diagnóstico4.
La presencia de quilomicrones en una muestra de plasma en ayunas es fácilmente observable. Justo después del muestreo de sangre y la centrifugación, se observará plasma lactescente (lechoso) cuando los TG sean ≥ 10 mmol/l (≥ 885 mg/dl), lo que indica la presencia de quilomicrones u otras lipoproteínas grandes ricas en TG. Adicionalmente, después del almacenamiento durante la noche a 4 °C se puede confirmar la presencia de quilomicrones, ya que solo estas lipoproteínas son capaces de formar una capa cremosa de sobrenadante en la parte superior del tubo, lo que se conoce como «prueba del refrigerador». Si la muestra no contiene VLDL, lo cual es típico en el FCS, la capa inferior del plasma estará clara2,5.
Los pacientes con TG en ayunas ≥ 10 mmol/l (≥ 885 mg/dl) en tres muestreos consecutivos deben evaluarse con un enfoque gradual. Una proporción entre TG totales y colesterol total > 5 mg/dl sugiere una gran cantidad de quilomicrones y VLDL circulantes. Para diferenciarse de la hiperlipidemia combinada poligénica, que se manifiesta con altos valores de TG y de lipoproteínas de baja densidad (LDL), en relación con un aumento de las concentraciones plasmáticas de Apo B-1007, se ha debatido sobre el uso de los valores de Apo B como herramienta de diagnóstico diferencial (FCS Apo B < 100 mg/dl y MCS Apo B > 130 mg/dl)4,7,15.
Se debe realizar una evaluación clínica para buscar las características físicas y las complicaciones del FCS. Deben excluirse factores secundarios que conducen a la elevación de los TG, como embarazo, obesidad, abuso de alcohol, diabetes no controlada, hipotiroidismo no tratado, síndrome nefrótico, mala alimentación, uso de diuréticos tiazídicos, estrógenos, corticosteroides, retinoides, resinas de ácidos biliares, antipsicóticos de segunda generación, bloqueadores beta y agentes antirretrovirales7. Estos factores secundarios, superpuestos a los trastornos de los TG leves subyacentes, a menudo exacerban la elevación marcada de los TG, pero en general responden al tratamiento y a los medicamentos que disminuyen los TG. Después de eso, se recomienda la secuenciación génica de LPL, LMF1, GPIHBP1, APOC2 y APOA5, que representa el método de referencia actual para el diagnóstico de FCS4.
Un panel de expertos clínicos europeos con experiencia en el diagnóstico de FCS se reunió en Amsterdam en julio de 2017 para discutir las estrategias óptimas para la identificación, el diagnóstico y la evaluación de estos pacientes4. Propusieron un diagnóstico pragmático para FCS, que podría ayudar a distinguir entre FCS y MCS, basado en ocho ítems biológicos/clínicos:
– Concentración de TG en plasma en ayunas > 10 mmol/l (> 885 mg/dl) en tres muestras consecutivas (+5 puntos).
– TG previos < 2 mmol/l (< 177 mg/dl) (−5 puntos).
– Falta de factores secundarios (+2 puntos) (excepto embarazo y anticonceptivos orales estrogénicos).
– Antecedentes de pancreatitis aguda (+1 punto).
– Sin antecedentes de hiperlipidemia combinada familiar (+1 punto).
– No respuesta a tratamiento hipolipemiante (disminución de TG < 20%) (+1 punto).
– Edad al inicio de los síntomas (< 40 años +1 punto, < 20 años +2 puntos, < 10 años +3 puntos).
– Dolor abdominal inexplicado (+1 punto).
Se dio un bajo peso al antecedente de pancreatitis, ya que no es específico para FCS frente a MCS, aunque parece ser más frecuente en el FCS. Además, una gran proporción de pacientes jóvenes con FCS aún no tienen antecedentes de pancreatitis y serían excluidos si el puntaje otorgara demasiado peso a este ítem. Los puntos de corte fueron > 10 puntos para FCS muy probable, < 9 puntos para FCS improbable y < 8 puntos para FCS muy improbable14.
Inicialmente probaron la puntuación FCS de los ocho ítems en dos cohortes de 29 pacientes en Roma y Lyon. La sensibilidad se estimó en una cohorte con FCS y la especificidad se probó en una cohorte con MCS. Teniendo en cuenta todos los pacientes estudiados, la sensibilidad general fue del 88% (intervalo de confianza del 95%: 0.97-0.76), con una especificidad general del 85% (intervalo de confianza del 95%: 0.94-0.75)14. Con respecto a este estudio, si bien puede ser fácilmente aplicable a cualquier población, los estudios de sensibilidad y especificidad deberían validarse en población latinoamericana para mejorar el abordaje de los pacientes con HTG grave, lo cual permitiría un diagnóstico más temprano al considerarse quién requeriría pruebas genéticas y por tanto un tratamiento oportuno.
Tratamiento
Antes de iniciar la terapia de reducción de TG se deben identificar y tratar las causas secundarias reversibles de HTG, como el hipotiroidismo, la diabetes mellitus tipo 2 mal controlada, la obesidad, la paraproteinemia, el lupus eritematoso sistémico, los trastornos renales y la anorexia nerviosa7. Además, los factores dietéticos que son causas establecidas de elevaciones de los TG, como el consumo excesivo de alcohol o de azúcar, requieren intervención. Cuando la HTG parece ser causada, al menos en parte, por medicamentos (como corticosteroides, tiazidas, betabloqueantes no selectivos, estrógenos, tamoxifeno, secuestrantes de ácidos biliares, ciclofosfamida, antirretrovirales y antipsicóticos de segunda generación)1,7 (Tabla 1), las opciones de tratamiento incluyen eliminar o disminuir la dosis del agente incitante, sustituyendo dentro de la misma clase de fármacos o cambiando por otro tipo diferente. Si estos cambios no son posibles o no son efectivos, puede ser necesario iniciar la farmacoterapia para reducir los TG1.
Tabla 1 Causas de hiperlipidemia secundaria
| Colesterol | Triglicéridos | HDL | |
|---|---|---|---|
| Hipotiroidismo | ↑↑ | ↑ | |
| Cushing | ↑ | ↑↑ | |
| Lipodistrofia | ↑ | ↑↑ | ↓ |
| Diabetes mellitus tipo 2 | ↑↑ | ↓ | |
| Síndrome nefrótico | ↑↑ | ||
| Insuficiencia renal crónica | ↑ | ↑ | ↓ |
| Colestasis | ↑↑ | ||
| Alcoholismo | ↑↑ | ↑ | |
| Embarazo | ↑↑↑ | ||
| Sepsis | ↑↑ | ||
| Bloqueadores beta | ↑ | ↓ | |
| Tiazidas | ↑ | ↑ | ↓ |
| Estrógenos orales | ↑↑ | ↑ | |
| Inhibidores de la proteasa | ↑ | ↓ |
HDL: lipoproteínas de alta densidad.
Modificaciones del estilo de vida
La restricción dietética de la ingesta de grasas es crucial, en especial para el tratamiento de la deficiencia homocigótica de LPL, es decir, el FCS. Las recomendaciones son reducir la ingesta de grasas en la dieta a menos del 20% de la ingesta calórica diaria total o incluso menos (10-15%). El consumo de grasa diario en gramos varía entre las diferentes revisiones, pero la mayoría coincide en que debe ser entre 20 y 40 g/día1,13. El éxito de la terapia depende de la aceptación individual de la restricción de grasas, incluidas las grasas saturadas y no saturadas. La adherencia puede ser un desafío clínico teniendo en cuenta que una dieta occidental normal contiene al menos 120 g de grasa por día. Los lactantes pueden recibir fórmulas con TG de cadena media, los cuales pueden usarse como una fuente de energía alternativa, ya que pueden absorberse directamente en la circulación portal debido a que no se incorporan a los TG ni a la formación de quilomicrones1,16.
Dado el papel central de los cambios en el estilo de vida, se debe ofrecer a los pacientes la oportunidad de recibir asesoramiento nutricional. Igualmente, es importante incrementar la actividad física a 2.5-5 horas por semana de ejercicio aeróbico de intensidad moderada. En los pacientes con sobrepeso, el objetivo es lograr la pérdida de peso, y en los pacientes con diabetes mellitus, lograr un buen control de la glucemia2.
Fibratos
Son hipolipemiantes útiles para reducir los TG elevados, ya que actúan sobre el receptor alfa activador del proliferador peroxisomal (PPAR-a), aumentan la actividad de la LPL y reducen la Apo C-III3, lo que incrementa la eliminación de lipoproteínas ricas en TG. Son una terapia de primera línea para la HTG grave y son efectivos en todos los pacientes con cualquier actividad residual de LPL. El manejo de la hiperlipidemia muestra una reducción en los episodios de pancreatitis paralela a su efecto en la reducción de los TG en hasta un 70%, aunque con una marcada variación interindividual1,16.
Como monoterapia, los fibratos probablemente reducen el riesgo de enfermedad cardiovascular ateroesclerótica. Muchas clínicas de lípidos en los Estados Unidos de América y Canadá consideran la terapia de fibratos en pacientes con alto riesgo de enfermedad cardiovascular ateroesclerótica y concentraciones persistentes de TG > 2.2 mmol/l (> 200 mg/dl), a pesar de alcanzar el objetivo de LDL y a pesar de las modificaciones en el estilo de vida1.
Estatinas
Las estatinas reducen los TG en proporción a su eficacia en la reducción de las LDL y a los valores basales de TG. Existen datos de que las estatinas reducen los TG de una manera proporcional a la dosis y sobre la eficacia obtenidos en un ensayo que reclutó pacientes con un valor de TG máximo de 8 mmol/l (708 mg/dl). Las estatinas actúan aumentando la expresión del receptor de LDL. El receptor de LDL puede unirse a dos ligandos: Apo B-100 y con menor afinidad Apo E. Las lipoproteínas ricas en TG tienen múltiples moléculas de Apo E y solo una Apo B-100, por lo que se eliminan preferentemente a pesar de su menor afinidad molecular por el receptor16.
Las estatinas a menudo no son efectivas para reducir significativamente los TG en los pacientes con FCS, lo que es una pista para el diagnóstico. Se observa un mayor efecto en pacientes con una elevación de los TG leve a moderada, que tienen la forma poligénica de HTG, en comparación con aquellos con FCS7. La terapia combinada de fibrato-estatina se usa en el tratamiento del déficit de LPL en función de sus mecanismos de acción complementarios, y en series de casos de hiperlipidemia de tipos IV y V se han observado reducciones de hasta un 80% en los TG, lo que se logra principalmente a través de acciones sobre el recambio de VLDL. La terapia combinada de clofibrato y lovastatina resultó en una disminución del 75% en los TG y una caída del 67% en el colesterol total en un estudio en pacientes con hiperlipidemia de tipo V16.
Niacina
La niacina tiene la capacidad de reducir los TG en un 20%, solo un poco menos que los fibratos, en pacientes con HTG moderada. La niacina se ha utilizado para tratar la HTG, ya que reduce la lipólisis periférica a través del receptor hidroxicarboxílico tipo 3, imitando el ligando fisiológico. La niacina disminuye la actividad de la diacil-glicerol-acil-transferasa hepática 2 (DGAT-2) implicada en el ensamblaje de las VLDL. Más recientemente se ha sugerido que su acción principal es reducir la cantidad de Apo C-III disminuyendo la actividad del PPAR-g coactivator-1 b (PGC-1b), que interactúa con la proteína de unión al receptor de esteroles 1c (SREBP-1c) y las vías PPAR-a involucradas en el metabolismo de los TG con una acción secundaria sobre la diacilglicerol O-aciltransferasa 1 (DGAT1) y la diacilglicerol O-aciltransferasa 2 (DGAT2). La niacina puede causar hiperglucemia y por tanto debe usarse con precaución en los pacientes con diabetes. Es aditiva a los fibratos en el tratamiento de la HTG. Hay pocos datos sobre el uso específico de la niacina en el FCS, pero los informes de casos muestran que tiene éxito en la reducción de los TG en la hiperlipidemia de tipo V con los efectos mediados por reducciones en la producción de quilomicrones (Apo B-48)16.
Omega-3
Los ácidos grasos insaturados omega-3 se manejan de manera diferente a los ácidos grasos saturados y reducen los TG en el plasma. Actúan parcialmente a través del receptor de ácido graso libre 4 (FFAR4) para aumentar la sensibilidad a la insulina y reducir la inflamación. Muestran propiedades antiinflamatorias y antiapoptóticas en líneas celulares pancreáticas. Las preparaciones modernas de ácidos grasos omega-3 altamente purificados que contienen ácido docosahexaenoico (DHA) o ácido eicosapentaenoico (EPA) reducen los TG de forma proporcional a la dosis en pacientes con HTG16. Las altas dosis de ácidos grasos omega-3 (> 1.5-2 g de etilo de icosapento más 1.2-1.5 g de etilo de doconexento al día) tienen un efecto reductor de los TG (alrededor del 25-30%). Sin embargo, la evidencia de los estudios que evalúan el uso de dosis bajas de ácidos grasos omega-3 (1 g al día) para prevenir eventos cardiovasculares es neutral. Por lo tanto, no hay razón para iniciar el tratamiento con dosis bajas de ácidos grasos omega-316.
Un informe científico de la American Heart Association ha abordado el uso de suplementos de ácidos grasos omega-3 para reducir el riesgo de enfermedad cardiovascular ateroesclerótica. Este informe se centra específicamente en el uso de estos agentes para tratar la HTG grave. Desde 2004, la Food and Drug Administration de los Estados Unidos de América ha aprobado varios tipos de agentes con fórmula de ácidos grasos omega-3 para el tratamiento de la HTG grave17.
Nuevos tratamientos
Tratamientos para reducir la Apo CIII
El volanesorsén es un oligonucleótido antisentido de segunda generación dirigido al ARN mensajero Apo CIII. Como ya hemos comentado, la Apo CIII reduce el aclaramiento de las lipoproteínas ricas en TG del plasma, a través de una vía diferente de la LPL, cuyo mecanismo no está del todo claro. Por lo tanto, se espera que la reducción de Apo CIII disminuya los TG plasmáticos al facilitar su eliminación. Los pacientes tratados con volanesorsén tuvieron una reducción de aproximadamente un 70% en los valores de TG después de 3 meses de tratamiento. Los efectos secundarios más comunes fueron reacciones en el lugar de la inyección, síntomas similares a la gripe, hipersensibilidad, efectos secundarios hepáticos y renales, y trombocitopenia18. En mayo de 2019, el volanesorsén fue aprobado en la Unión Europea para el tratamiento de pacientes adultos con FCS basándose en los resultados positivos de los estudios multinacionales de fase III APPROACH y COMPASS18,19.
Inhibición de la diacilglicerol aciltransferasa
El pradigastat y el AZD7687 son inhibidores orales de la DGAT1 intestinal, que regula la absorción de grasas en la dieta y la síntesis de TG1. El pradigastat mostró eficacia para reducir los TG en pacientes con FCS. Se evaluó en un estudio abierto con seis pacientes con FCS y mostró una reducción del 72% en los valores de TG con una dosis de 40 mg. El estudio con 45 pacientes con FCS aleatorizados mostró buena seguridad y tolerabilidad, pero el 72% de los participantes informaron diarrea. Esto podría complicar el uso adicional de pradigastat7.
Inhibidores de la secreción de quilomicrones y VLDL
El déficit de LPL se relaciona con la eliminación de partículas de quilomicrones y VLDL. Por lo tanto, las terapias que interfieren con la síntesis de partículas se han considerado en el FCS. La lomitapida es un inhibidor de la proteína de transferencia de TG microsomales con licencia para la hipercolesterolemia familiar homocigótica, en la que logra una reducción del 50% de LDL y una reducción paralela más pequeña de los TG. Su uso en el FCS se ha explorado en un caso basándose en un protocolo en el que la terapia con lomitapida disminuyó los TG en un 75% y redujo los ingresos por pancreatitis durante un periodo de 13 años. Sin embargo, el tratamiento se asoció con el desarrollo posterior de esteatohepatitis y fibrosis hepática16.
Terapia génica
La terapia de reemplazo del gen, denominada Alipogene Tiparvovec, es un vector viral adenoasociado no replicativo del serotipo 1 que fue diseñado para administrar y expresar el gen LPL. El medicamento fue administrado por inyección intramuscular. Los efectos de la terapia fueron de corta duración, pero se observó una menor incidencia de pancreatitis en los pacientes con FCS hasta 6 años después del tratamiento7. Una comparación de las tasas de ingreso por pancreatitis anteriores y posteriores al tratamiento indicó una reducción del 50% en el número y posiblemente en la gravedad de los eventos de pancreatitis después de 2 años. Alipogene tiparvovec fue autorizado por la Agencia Europea de Medicamentos, pero las malas perspectivas comerciales y las incertidumbres sobre el reembolso provocaron su retirada en el año 201716.
Metas de tratamiento
Existe el riesgo que al elevarse las concentraciones de TG se incremente el riesgo de pancreatitis, pero es difícil establecer un umbral de tratamiento específico. Los objetivos del tratamiento son controvertidos, ya que hay poca evidencia sistemática para el tratamiento del FCS. Cualquier base de evidencia disponible está relacionada con el riesgo de enfermedad cardiovascular y no con la pancreatitis. Muchos médicos considerarían como deseables unas concentraciones de TG < 10 mmol/l (< 885 mg/dl) y como ideales < 5 mmol/l (< 400 mg/dl). Sin embargo, los organismos reguladores y los estudios comerciales apuntan a objetivos de TG establecidos a partir de estudios sobre enfermedad cardiovascular o síndrome metabólico, y típicamente informan un objetivo de TG < 2.3 mmol/l (< 200 mg/dl) o < 1.7 mmol/l (< 150 mg/dl). No existe un objetivo para los TG en ninguna guía reciente de enfermedad cardiovascular, pero el riesgo de esta se encuentra incluido en las recomendaciones para la reducción del colesterol no unido a lipoproteínas de alta densidad a < 2.5-3.0 mmol/l (< 100-120 mg/dl)16.
Pancreatitis aguda secundaria a hipertrigliceridemia
La HTG es la tercera causa de pancreatitis aguda y puede llegar al 10% de todos los casos, elevándose hasta el 50% durante el embarazo. La gravedad y las complicaciones tienden a ser mayores, y por lo tanto el tratamiento médico pertinente es fundamental. La pancreatitis aguda secundaria a hipertrigliceridemia es una enfermedad que se presenta generalmente en pacientes con valores de TG > 11.3 mmol/l (1000 mg/dl)20. El riesgo de pancreatitis aguda en los pacientes con TG > 11.3 y 22.5 mmol/l (>1000 y >2000 mg/dl) es del 5% y el 10-20%, respectivamente. Sin embargo, este umbral es arbitrario, la cifra de TG para que se presente la pancreatitis se desconoce y la gravedad no parece ser directamente proporcional a la cantidad de TG21.
El tratamiento agudo es la clave del manejo, ya que el 50% de la mortalidad se observa en este tiempo, y es similar al manejo de la pancreatitis aguda de otras causas20. El tratamiento de la pancreatitis aguda en pacientes con HTG grave incluye medidas básicas, como ayuno estricto y manejo hídrico2. La interrupción de la ingesta de alimentos también se utiliza para detener la producción de lipoproteínas secretadas posprandialmente desde el intestino. En general, los TG plasmáticos se reducen a la mitad cada 48 horas en esta situación16.
Por el momento no hay un tratamiento específico para la HTG grave, pero se han utilizado diferentes opciones terapéuticas, como la administración de insulina, heparina y plasmaféresis, para disminuir las concentraciones plasmáticas de TG20.
En casos agudos muy graves, el exceso de lipoproteínas ricas en TG puede eliminarse físicamente mediante plasmaféresis. El objetivo del tratamiento es lograr las concentraciones de TG más bajas posible7. La plasmaféresis disminuye agudamente los TG en un 50-80%, y por lo tanto, en teoría, puede eliminar el factor causante en la pancreatitis inducida por HTG. Sin embargo, una serie de reportes de 22 casos de pancreatitis aguda con valores medios de TG de 45 mmol/l (3982 mg/dl) mostró unas reducciones de TG comparables, alrededor del 70% en 48 horas, utilizando un tratamiento conservador, sin plasmaféresis. Los valores de TG aumentan rápidamente después de la plasmaféresis si la causa no se maneja de manera adecuada1.
La insulina promueve la síntesis y la activación de la LPL, disminuyendo los TG e incrementando la degradación de quilomicrones. Se utiliza en diferentes esquemas de infusión dependiendo de si el paciente tiene diabetes, logrando reducir los TG a < 11.3 mmol/l (1000 mg/dl) en un periodo de 3-4 días de infusión. En una serie de 10 casos, el uso de insulina intravenosa se relacionó con un descenso del 40% de los TG en 24 horas en pacientes con HTG grave y con un descenso del 87% en los pacientes tratados con insulina más ayuno. La terapia con insulina ha mostrado ser segura y efectiva en el tratamiento de la pancreatitis aguda secundaria a hipertrigliceridemia20.
La heparina, en el contexto de la pancreatitis aguda secundaria a hipertrigliceridemia, altera el proceso inflamatorio por su acción sobre las células endoteliales, bloqueando la adhesión plaquetaria y de leucocitos al endotelio y reduciendo el reclutamiento leucocitario, llevando a una inhibición de la respuesta inflamatoria, reduciendo la mortalidad y mejorando la puntuación de Balthazar. Después de la interrupción de la heparina puede presentarse HTG de rebote, por lo que se recomienda usar heparina después del manejo inicial basado en ayuno, terapia hídrica e infusión de insulina; por lo anterior, la heparina no sería una terapia de elección como monoterapia20.
Conclusión
La HTG grave es diagnosticada como hallazgo incidental en la mayoría de los casos. Dentro de sus causas, el FCS está subdiagnosticado y subtratado, por lo cual se debe conocer sobre él. Diferenciar el FCS del MCS es importante para establecer las posibles complicaciones que se pueden presentar, realizar un diagnóstico genético oportuno y orientar el manejo más apropiado. Para ello, consideramos necesario que el personal médico conozca sobre la existencia de esta enfermedad, se realicen estudios para evaluar la epidemiología al respecto y se validen los puntajes de diagnóstico.

