











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
La cardiomiopatía hipertrófica apical (ApCMPH) es una forma poco común de cardiomiopatía hipertrófica (CMPH) que se caracteriza por un compromiso casi exclusivo del ápex1, y en ella se visualiza una morfología típica en as de picas tanto en la ventriculografía como en la resonancia magnética cardiaca. La prevalencia de CMPH es del 0.2% y es similar entre los diferentes grupos raciales; sin embargo, la forma apical es más común en población asiática (15%) en comparación con la de los Estados Unidos de América (2%)2,3.
La ApCMPH, a pesar de haber sido descrita por primera vez hace mas de 40 años, sigue siendo un desafío diagnóstico para el personal médico. A través de esta presentación de caso, se hace una revisión de la literatura sobre el tema con el fin de identificarla y comprenderla con mayor facilidad.
Presentamos el caso de un paciente octogenario que debutó con síntomas de dolor precordial típico y síncope cardiogénico. Los hallazgos electrocardiográficos alarmaron al personal de atención inicial y generó una búsqueda que terminó en un diagnóstico no esperado y poco frecuente4.
Caso clínico
Varón de 82 años con antecedentes personales de hipertensión arterial y tabaquismo, sin historia familiar relevante. El paciente consulta a un hospital de primer nivel en su localidad por un cuadro clínico de 2 horas de evolución consistente en dolor torácico opresivo retroesternal, asociado a un episodio de síncope. En la evaluación inicial realizaron un electrocardiograma (ECG) (Fig. 1), que mostró inversión de las ondas T en precordiales (hasta de 18 mm) con depresión del segmento ST en V2-V4, razón por la que sospecharon infarto agudo de miocardio, iniciaron manejo farmacológico para este y remitieron al paciente a una institución de tercer nivel. En el interrogatorio dirigido, el paciente refiere que desde hace 2 años ha presentado varios episodios de síncope a repetición, por los cuales nunca consultó. Niega angina de esfuerzo o disnea. Con la alta sospecha del personal de salud de infarto agudo de miocardio sin elevación del ST, se decide ingresar al paciente a coronariografía, donde se observan las arterias coronarias sin lesiones significativas, hallazgo que sorprende al personal médico. Se procede a realizar ventriculografía izquierda, en la cual se evidencia una hipertrofia grave del ventrículo izquierdo, con compromiso de los segmentos medios ventriculares y apicales, generando la apariencia angiográfica clásica de as de picas (Fig. 2), con lo que se confirmó el diagnóstico de ApCMPH.
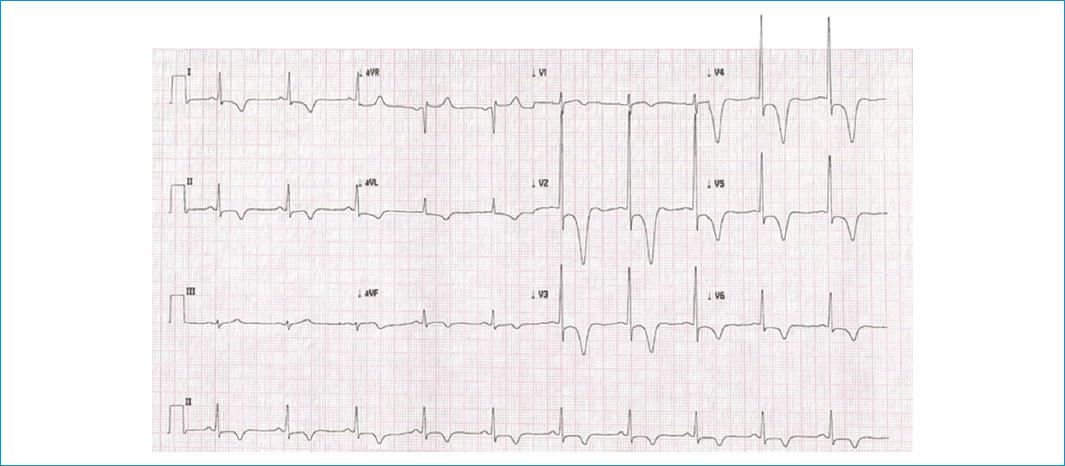
Figura 1 Electrocardiograma en el cual se observa inversión de ondas T en todas las precordiales (de hasta 18 mm) con depresión del segmento ST en V2-V4.
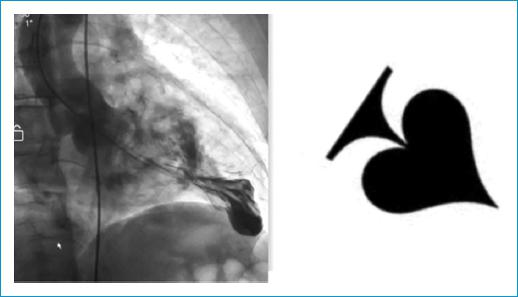
Figura 2 Ventriculografía que evidencia el ventrículo izquierdo muy hipertrófico, en particular en los segmentos medio ventricular y apical, generando una apariencia angiográfica de as de picas característica de la cardiomiopatía hipertrófica apical. Adicionalmente se observa un aneurisma apical
La ecocardiografía transtorácica que se realizó mostró una hipertrofia apical con máximo grosor a este nivel, de 19 mm, la cual produce un colapso del ápex ventricular izquierdo, y se documentó un gradiente sistólico máximo de 46 mmHg (Fig. 3).
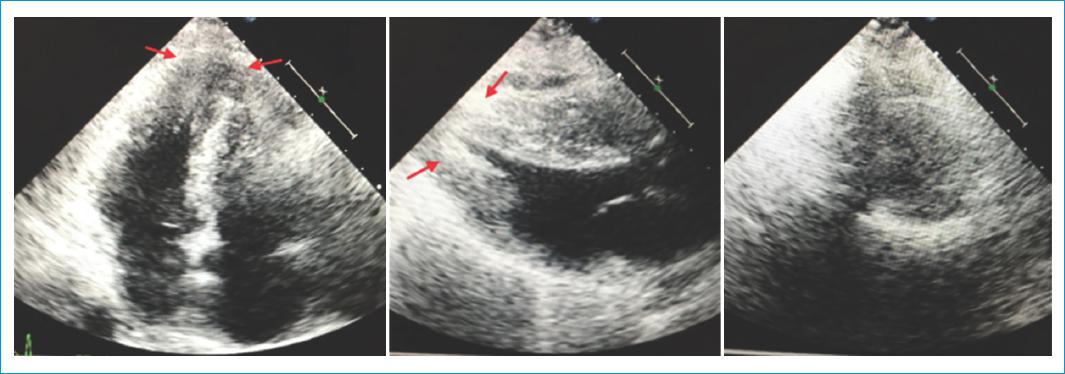
Figura 3 Ecocardiografía que evidencia una hipertrofia de los segmentos medio y apical (flechas) con disminución significativa de la luz, la cual produce colapso del ápex ventricular. De izquierda a derecha: imagen apical de cuatro cámaras, eje largo paraesternal y eje corto a nivel apical.
Finalmente se realizó un monitoreo Holter de 24 horas que evidenció episodios de taquicardia ventricular no sostenida. Por los hallazgos clínicos y del Holter se implantó un cardiodesfibrilador como prevención primaria. El paciente fue dado de alta de la institución con beta-bloqueadores de forma ambulatoria e indefinidamente.
Discusión
Se denomina cardiomiopatía la enfermedad que afecta primariamente al musculo cardiaco y que, con independencia de su etiología, se manifiesta por dilatación, hipertrofia o restricción al llenado del corazón sin que esté presente una sobrecarga hemodinámica. Por otro lado, la hipertrofia miocárdica constituye un mecanismo compensador fisiológico que el corazón utiliza para compensar sobrecargas de trabajo cardiaco; si el corazón no está sometido a ninguna sobrecarga hemodinámica (como podría ser una hipertensión arterial sistémica) y aparece hipertrofia de sus paredes, el proceso se debe a la presencia de una enfermedad primaria del miocardio que se manifiesta por hipertrofia patológica, denominada CMPH5.
La ApCMPH, es una variante poco frecuente en la cual la hipertrofia se localiza principalmente en el ápice ventricular izquierdo. Las primeras descripciones fueron en población asiática, y desde entonces la mayoría de los reportes son en esta población, llegando a constituir entre el 15% y el 25% de todas las CMPH en Japón6.
Tiene herencia familiar con un patrón autosómico dominante, aunque también existen formas esporádicas de la enfermedad, causadas por mutaciones adquiridas del gen de la proteína del sarcómero7. Los hallazgos histológicos característicos incluyen edema, fibrosis, desorganización y núcleos extraños en el miocardio8. Es más frecuente en el sexo masculino y en edades de 40 años en promedio. Llama la atención que nuestro paciente era octogenario9.
Morfológicamente, la ApCMPH se puede dividir en tres tipos: focal puro, difuso puro y mixto; de ellos, el focal puro es el más común, pero en la práctica clínica esta subclasificación no se utiliza ampliamente y se desconoce su relevancia clínica10.
Manifestaciones clínicas
Un gran porcentaje de los pacientes cursan completamente asintomáticos y el diagnóstico se hace por hallazgos incidentales en el ECG. En otros casos podría confundirse con un síndrome coronario agudo, como ocurrió en el paciente descrito, por cursar con dolor torácico asociado a anomalías en la repolarización en el ECG11, en ausencia de enfermedad coronaria epicárdica. El dolor de la ApCMPH en el pecho está causado por isquemia miocárdica debido al estrechamiento de los pequeños vasos coronarios intramurales en el miocardio hipertrofiado, así como a una mayor demanda de oxígeno miocárdico por hipertrofia de miocitos12. Otras manifestaciones clínicas descritas son síncope, disnea de esfuerzo, palpitaciones, mareo, fatiga y síntomas de insuficiencia cardiaca13; estos últimos debido al llenado deficiente del ventrículo izquierdo secundario a la rigidez de la pared y la consiguiente disminución del gasto cardiaco que puede provocar insuficiencia cardiaca diastólica14.
Diagnóstico
La aproximación inicial se realiza mediante un ECG de 12 derivaciones, en el que se podrían encontrar alto voltaje QRS, anomalías de la repolarización y las inversiones clásicas de las ondas T profundas en las derivaciones precordiales, observadas en el 93% de los pacientes. Las ondas T negativas gigantes, definidas como un voltaje ≥10 mm, se encuentran en menor porcentaje (47% de los pacientes), como se puede apreciar en el ECG del paciente descrito, con ondas T negativas de hasta 18 mm. Otras características clásicas son hallazgos de hipertrofia ventricular izquierda (alrededor del 65% de los casos)1,13,15,16.
Posterior a la sospecha diagnóstica con los hallazgos previos, se procede a la demostración de la hipertrofia apical. Las ayudas diagnósticas no invasivas más útiles son la ecocardiografía bidimensional y la resonancia magnética cardiaca. La ecocardiografía bidimensional suele ser el estudio diagnóstico inicial. Los criterios diagnósticos incluyen hipertrofia del ventrículo izquierdo asimétrico, limitada al vértice del ventrículo izquierdo, con un grosor de la pared apical ≥ 15 mm (en nuestro caso 19 mm) y una relación de grosor apical máximo a la pared posterior ≥ 1.5 mm. La detección de la ApCMPH puede ser difícil por este método, en especial cuando la calidad de la imagen del ecocardiograma transtorácico es subóptima, por limitación para visualizar el ápice. El uso de ecocardiografía de contraste aumenta el rendimiento del estudio16,17.
La resonancia magnética cardiaca, por su parte, puede obtener imágenes en cualquier plano con una mayor resolución y apreciación de la morfología cardiaca, por lo que determina de manera confiable la estructura del ápice cardiaco. En una serie reciente de ApCMPH, nueve de 105 pacientes tenían ecocardiogramas negativos, que en la revisión después de la resonancia magnética cardiaca fueron positivos18.
Por otro lado, la ayuda diagnóstica invasiva utilizada es la ventriculografía, la cual muestra una configuración distintiva del ventrículo izquierdo con forma de as de picas5, lo que ha sido típicamente descrito. En el caso presentado, dada la alta sospecha de infarto de miocardio sin elevación del ST, el paciente fue llevado a coronariografía inmediatamente tras llegar al centro de atención de tercer nivel, encontrándose coronarias normales, por lo que se procedió, entonces, a realizar una ventriculografía y se llegó finalmente al diagnóstico de ApCMPH.
Pronóstico y seguimiento
A diferencia de otras variantes de CMPH, el pronóstico de la ApCMPH es relativamente benigno; sin embargo, un tercio de los pacientes pueden experimentar complicaciones potencialmente mortales, como infarto agudo de miocardio, infarto apical con aneurisma, taquicardia ventricular, muerte súbita de causa cardiaca o accidente vascular cerebral16,17.
Los factores de mal pronóstico son la edad avanzada, la presencia de hipertensión o diabetes, la fibrilación auricular basal y los hallazgos en la resonancia magnética cardiaca de aneurisma apical y extensión del realce tardío con gadolinio13. El paciente aquí descrito contaba con múltiples factores de mal pronóstico (edad avanzada, hipertensión arterial y presencia de aneurisma apical, visualizado en la ventriculografía).
En un seguimiento de 193 pacientes durante una mediana de 78 meses se encontró que las mujeres tenían mayor incidencia de insuficiencia cardiaca, fibrilación auricular y muerte que los hombres, y la supervivencia en general de los pacientes observados a 20 años fue del 47%. Dados los limitados datos disponibles debido a la baja prevalencia, está indicado un seguimiento periódico de por vida, incluso en pacientes asintomáticos13.
¿Dispositivos implantables?
Ninguna guía define las indicaciones exactas para implantar un desfibrilador en pacientes con ApCMPH, pero el consenso de expertos apoya el uso de un desfibrilador implantable para la prevención primaria en pacientes seleccionados de alto riesgo, especialmente en aquellos con uno de los cinco factores de riesgo siguientes: historia familiar de muerte súbita de causa cardiaca, síncope inexplicable, episodios de taquicardia ventricular no sostenida asintomática, respuesta de presión arterial anormal al ejercicio y grosor de la pared ventricular izquierda > 30 mm11,19. En nuestro caso, se decidió el implante del dispositivo principalmente porque el paciente contaba con dos de los factores: taquicardia ventricular no sostenida en el monitoreo Holter realizado y síncope inexplicable; y además presentaba otros factores de mal pronóstico, ya descritos.
Tratamiento
El manejo de la ApCMPH se basa en tres pilares fundamentales: control de los síntomas, pruebas genéticas y prevención de secuelas cardiovasculares adversas20. Los beta-bloqueadores son la terapia inicial recomendada en los pacientes con ApCMPH para tratar la angina al disminuir el oxígeno del miocardio, prevenir arritmias ventriculares no sostenidas y disminuir la frecuencia cardiaca para mejorar el llenado diastólico. Los resultados a largo plazo de la terapia con beta-bloqueadores en los pacientes con ApCMPH son desconocidos y se necesitan más estudios. Los bloqueadores de los canales de calcio no dihidropiridínicos son una terapia de segunda línea si los pacientes no pueden tolerar los beta-bloqueadores14,16.
Es importante controlar la comorbilidad, en especial la hipertensión y la enfermedad valvular, que funcionan como factores estresantes mecánicos, para prevenir una hipertrofia ventricular patológica adicional21. El manejo adicional de la ApHCM incluye la colocación de un desfibrilador implantable en los pacientes de alto riesgo, pruebas genéticas tanto para el paciente como para los familiares de primer grado, y un examen electrocardiográfico y ecocardiográfico de pacientes y familiares de primer grado. De acuerdo con las directrices del American College of Cardiology y la American Heart Association para la CMPH, las recomendaciones para el cribado genético y el seguimiento son similares tanto para la ApCMPH como para la CMPH obstructiva. Los familiares de primer grado que dan positivo para el genotipo, pero no muestran el fenotipo en el examen inicial, deben someterse a ecocardiografía y ecocardiografía seriadas: cada 3-4 años los adultos y cada 12-18 meses los niños. Los miembros de la familia que son genotipo negativos no necesitan más seguimiento14.
Conclusiones
La ApCMPH es una enfermedad poco frecuente en nuestro medio, de la cual no se tiene tanto conocimiento como de otras formas de CMPH, debido a su baja prevalencia. Sin embargo, cada vez se reporta mayor número de casos en poblaciones no asiáticas. Por lo anterior, es importante conocerla e incluirla entre las posibilidades diagnósticas en la evaluación inicial del dolor torácico típico, así como otras variantes de la CMPH, ya que el manejo adecuado puede prevenir secuelas devastadoras.

