











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
PermalinkIntroduction
Scimitar syndrome (SS) or venolobar syndrome is a congenital malformation consisting of total or partial anomalous drainage of the right pulmonary veins, more frequently to the inferior vena cava and to a lesser extent to the right atrium, left atrium, azygos vein, or suprahepatic veins1,2; regularly associated with hypoplasia of the right lung and the right bronchial arterial tree3,4. Some authors only consider the syndrome when the abnormal drainage occurs to the inferior vena cava, otherwise, they classify it as a variant of SS5. It was described by Cooper for the 1st time in 1836 after the autopsy of an infant6 and then named by Neill in 19607. SS constitutes 0.5-2% of congenital heart disease (CHD), 3-5% of cases of abnormal return of pulmonary veins and it has been described in 3-6% of the patients with partial abnormal venous connections8-11. There is an estimated incidence of 1-3 per 100,000 live births, with a female/male ratio of 2-18,12.
The age of diagnosis varies according to the severity of the symptoms, which is proportional to the degree of cardiac and pulmonary formation. To date, the highest reporting age has been associated with mild structural alterations13. The case of a patient with a late diagnosis of the syndrome is reported, despite severe pulmonary and cardiac alterations.
Clinical case
A 76-year-old woman from Manizales with a history of dextroposition diagnosed at age 30, at age 74, she was diagnosed with hypothyroidism and hypertension. She was admitted to the institution with a 6 months hystory of dyspnea with progressive worsening until minimal effort, concomitantly cough with hyaline expectoration and subjective fever; extension paraclinical tests with transesophageal echocardiography that reported severe pulmonary hypertension (PHT) 96 mmHg secondary to congenital shunt type atrial septal defect, dextroposition, and abnormal communication between the right pulmonary veins and the inferior vena cava, compatible with SS, confirmed by CT angiography (Figs. 1-2), which reported concomitant lung infection.
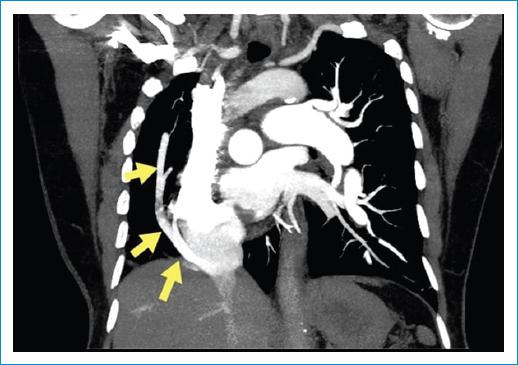
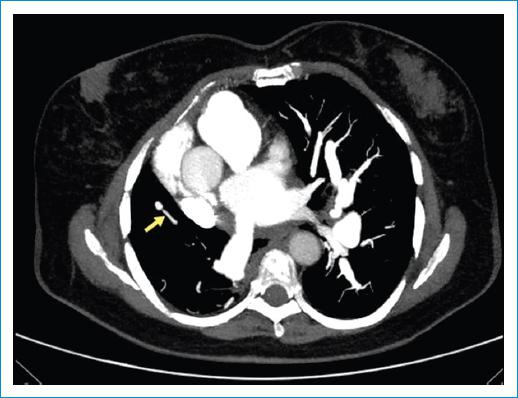
The patient consults 2 months later due to persistent dyspnea, despite the prescribed treatment, reporting symptoms compatible with pulmonary superinfection; outpatient spirometry report with a restrictive pattern that does not improve with a bronchodilator. Piperacillin tazobactam is administered and to optimize management. For severe PHT, a right catheterization was requested, which reported pulmonary arterial pressure 61/27, mean arterial pressure 40 mmHg, pulmonary vascular resistance 6.8 Wood units, pre-capillary PHT with a transpulmonary gradient of 30, pulmonary diastolic gradient of 17, RVP/SVR ratio 0.25, wedge pressure 10, and no secondary LV dysfunction; plethysmography was performed in which increased total pulmonary resistance was documented with an expiratory reserve volume of 0.76 OL (148%), total lung capacity 5.75 OL (128%), residual volume 3.85 (187%), RV/TLC ratio 66.9 (148%), and specific airway resistance 14.5. Once antibiotic management was completed, a poor response to management with bosentan was considered, so ambulatory management was changed to riociguat, with a better clinical response, to date without new consultations.
Discussion and review of the literature
The SS; also known as Halasz syndrome, mirror image lung syndrome, hypogenetic lung syndrome, right peribronchial pulmonary artery syndrome, and bronchovascular vena cava syndrome12; takes its name from the eastern saber with a long curved blade that resembles the shape of the drainage visible on the chest X-ray, known as the scimitar sign6,14. Cardiovascular diseases, pulmonary hypoplasia, and dextrocardia or dextroposition have been reported as the most prevalent concomitants, which are more severe at a younger age of diagnosis so that only mild forms of the disease have been described in adults3.
The exact process that contributes to the development of SS is not currently known. However, it is estimated to be due to an embryological error in lung development in early embryogenesis9. To date, it has not been established as a hereditary syndrome and specific genes have not been implicated, however, in a recent article, a family case was described in which genetic analysis revealed a deletion of chromosome 15q 15.3 that has been associated with the syndrome of deafness-infertility secondary to alterations in the STRC and CATSPER2 gene, raising for the 1st time the possibility of finding autosomal dominant inheritance; however, complete sequencing is pending15.
Some authors made a hypothesis pathogenetic, consisting of a developmental alteration in Streeters stage XIV (28-30 days of embryonic development), when the common pulmonary vein of the left atrium connects with the pulmonary venous sinus, at which time involution of the primary venous connections. However, the primitive canal of the primary pulmonary venous connection that empties into the suprahepatic portion of the inferior vena cava remains patent16.
The right pulmonary artery hypoplasia is thought to lead to ipsilateral pulmonary hypoplasia, due to decreased blood flow17. However, in another article, a hypothesis was raised based on the processes of lung development, which are inducing epithelium-mesenchymal interactions, growth, cell differentiation, movement, and apoptosis18. The first is responsible for the construction and differentiation of the bronchial tree and the establishment of the respiratory epithelium; it is believed that when this process is altered, it causes dysplasia and hypoplasia of the organ in the SS16. In addition, there is a case report in which the right pulmonary hypoplasia precedes the hypoplasia of the pulmonary artery, for which the mechanism is considered to be bidirectional17.
Some anatomical variants of the syndrome have been described; the first is called “left variant,” characterized by aortopulmonary collateral circulation to the left lung, with various degrees of hypoplasia of both the left lung and the pulmonary artery and at least one anomalous vein that drains to the inferior vena cava5. The second consists solely of the presence of an anomalous right pulmonary vein that drains to the inferior vena cava, without hypoplasia of the right lung or pulmonary artery. The third variant is given by the three characteristics mentioned in the first, together with a pulmonary venous drainage that can be normal or abnormal, and that goes to a different site from the inferior vena cava, the least frequent being the azygos vein5,19. Concomitantly associated heart defects have been described, the most common being atrial septal defect (80%), patent ductus arteriosus (75%), ventricular septal defect (30%), and pulmonary vein stenosis (20%); however, tetralogy of Fallot, hypoplasia or coarctation of the aortic arch, and hypoplastic left heart syndrome can also be found9.
It is classified into three groups according to age and severity of symptoms4. Group I, called “isolated adult,” constitutes the form in adults, they are typically acyanotic, asymptomatic; the diagnosis is generally made as an incidental finding or by the development of symptoms years or decades after birth in association with heart failure or cirrhosis20; they may present with a small atrial septal defect and do not develop pulmonary arterial hypertension12,14,21. Group II is characterized by its association with complex CHD, within which atrial septal defect and patent ductus arteriosus are excluded, it is known as “childhood CHD”; it is diagnosed in the neonatal period and presents with respiratory failure; it is commonly associated with PHT and due to its severity it frequently requires surgical treatment22,23. Group III is the “isolated infantile” form, most frequently diagnosed in childhood, it begins with recurrent respiratory infections that generally affect the right lower lobe, the severity and frequency of infections are related to the degree of hypoplasia and PHT; it has a poor prognosis as it is commonly characterized by severe PHT12,14,21,24.
Some authors have suggested that the partial anomalous pulmonary venous connection becomes clinically significant only when 50% or more of the pulmonary blood flow returns abnormally14,25. In the bibliographic search carried out, few cases described in adulthood were found, all with mild cardiac anomalies, among which a 57-year-old woman and two 68- and 84-year-old men stand out for being the longest-lived identified13,26,27. No case was found with significant cardiac abnormalities in adulthood (dextroposition, atrial septal defect, or severe PHT), such as the patient reported in this case. It should be noted that in pregnant women with a de novo diagnosis, it forces the end of the pregnancy early28.
The reasons for consultation in adulthood have been identified as dyspnea on exertion and recurrent respiratory infections; with a finding on physical examination of a precordial murmur12,29. In infants, the condition usually appears with tachypnea, cyanosis, growth failure, and PHT30.
PHT and the degree of pulmonary hypoplasia are recognized as the causes of the severe symptoms and poor prognosis of the syndrome, which can lead to premature death14,23,31-33. A retrospective study with an average follow-up of 4.2 years with 80 patients found that the only independent risk factor associated with mortality is PHT34. The major contributor to this is the large left-to-right shunt through the anomalous pulmonary vein or from the systemic arterial supply to the right lung35. The shunt from left to right in adults is <50% in 82% of patients, in those who present it, more than 50% develop dyspnea, chest infections, and PHT36. If the shunt is large enough, the right heart failure can occur37.
Diagnosis
Clinical suspicion in adulthood arises in the presence of exertional dyspnea, exercise intolerance, or a chest murmur. A routine chest X-ray should be performed, where the scimitar sign may raise diagnostic suspicion. However, this occurs in 70% of cases in adolescence or adulthood and is usually absent in infants38; it has also been reported in other vascular collaterals, so this sign is considered insufficient for its diagnosis11,38. Other findings may include dextrocardia and volume loss in the hemithorax with compensatory hyperinflation of the abnormal lung12.
Early diagnosis and appropriate treatment can improve the prognosis of patients31. Echocardiography is the technique of choice for the diagnosis of CHD, but it has several limitations in the detection and evaluation of pulmonary venous connections14,39. Tomography, for its part, offers the possibility of non-invasive and rapid acquisition with high resolution, its main disadvantage being the use of ionizing radiation14,38-40.
Cardiac catheterization and angiography are the most useful diagnostic studies to confirm SS, but they are not always necessary. They provide pulmonary vascular resistance, degree of left-to-right shunting, anomalous vein anatomy, grade of PHT, pulmonary artery anatomy, associated heart defects, and may demonstrate additional collateral systemic arteries from the thoracoabdominal aorta to the lung6,12,29,41. However, it may not reveal the anatomical details of small accessory or anomalous vessels36. Images with three-dimensional reconstruction clearly depict abnormal pulmonary venous structures, with detection rates approaching 100%39.
Treatment
Treatment is carried out according to the age of diagnosis, the number, and the location of the anomalous veins. The infantile form tends to show signs of heart failure, so medical therapy should be started immediately; according to the persistence of the symptoms, angiography and obliteration of the abnormal systemic arteries can be performed using a catheter. This allows for clinical improvement by reducing abnormal flow and pulmonary artery pressure. However, if the patient has associated cardiac defects and is symptomatic or the pulmonary-to-systemic flow ratio is greater than 1.5 in asymptomatic patients, they benefit from surgical correction23,32,34.
A relationship between a lower age at diagnosis with severe symptoms as an indicator of a very poor prognosis and an older age at diagnosis with a lower incidence of complications in surgical management and better survival is reported29,38.
Surgical treatment can be achieved either by resecting the lung with abnormal venous drainage or by performing a surgical correction for flow redistribution9. Cardiopulmonary bypass methods that have been used include direct or indirect anastomosis of the scimitar vein in the posterior part of the left atrium when the atrial septum is intact, or division and reimplantation of the abnormal pulmonary vein in the right atrium, sometimes requiring the use of grafts. If the stenosis presents as a complication, dilation can be performed with a percutaneous balloon, with good hemodynamic and symptomatic improvement42.
A multicenter study of the demonstrated that corrective surgery can be performed safely with low mortality and morbidity, regardless of the type of surgical technique used, especially if it is performed before the development of PHT42, thus the optimal moment to the intervention is preschool age12,14,42.
In other studies, it has been reported that patients who have developed mild-to-moderate PHT, surgical repair is generally safe and effective, although medical and catheter-guided therapies may be safer. Finally, in patients who have already progressed to severe PHT, a lung transplant may be necessary12,32,42.
Lobectomy or pneumonectomy is only indicated if there are recurrent infections, diffuse bronchiectasis, persistent hemoptysis, or marked hypoplasia of the right lung21,21,40,43.
Conclusion
SS is a rare entity with a poor prognosis for life when it develops in conjunction with dextrocardia or dextroposition and PHT, the latter can be considered as the main cause of complication and predisposition to recurrent chronic and acute inflammatory lung diseases and its progression should be studied as a multicausal phenomenon.
The unusual presentation of the reported clinical case is highlighted given the age of the patient, mild symptoms during most of her life, associated with structural alterations of poor prognosis such as left to right flow, severe PHT, dextroposition, with restriction and decrease of lung capacity documented in paraclinical patients, a course not described to date in the scientific literature which highlights the importance of early diagnostic suspicion even in atypical presentations, and optimal medical management to control symptoms and PHT.

