











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
Amiloidosis es un término que se refiere a un conjunto de enfermedades que se producen por la infiltración y depósito de proteínas plegadas anormalmente en los espacios extracelulares de diversos órganos, que captan específicamente la tinción rojo Congo; poseen una ultraestructura fibrilar y patrón de láminas con plegamiento beta. El subtipo particular de la enfermedad está determinado por el tipo de proteína y el órgano en el cual se depositen y de acuerdo a esto serán las manifestaciones clínicas que presente. Actualmente, se sabe que existen más de 32 proteínas diferentes (y muchas más variantes), así como casos de amiloidosis asociadas a enfermedades inflamatorias crónicas, y continuamente se agregan tipos de proteínas adicionales a esta lista; sin embargo, el 98 % de los casos se debe al depósito de fibrillas compuestas de la replicación monoclonal de la cadena liviana de inmunoglobulinas (amiloidosis AL) o de transtiretina (amiloidosis ATTR); esta última, a su vez, puede ser una forma hereditaria o salvaje1.
Caso clínico
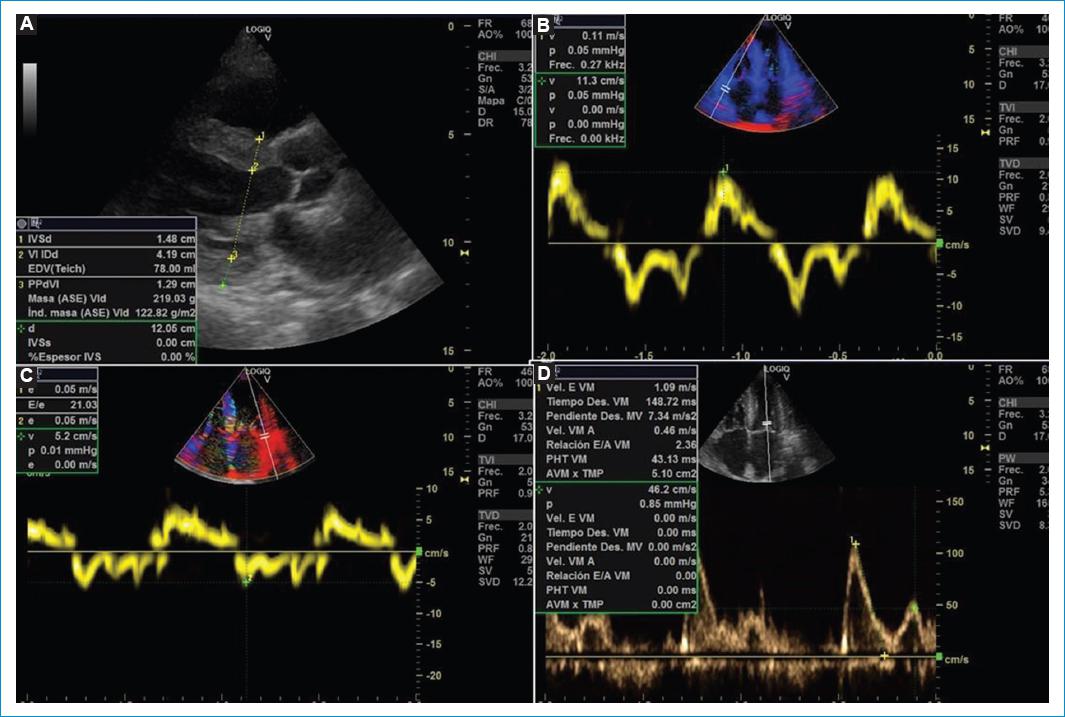
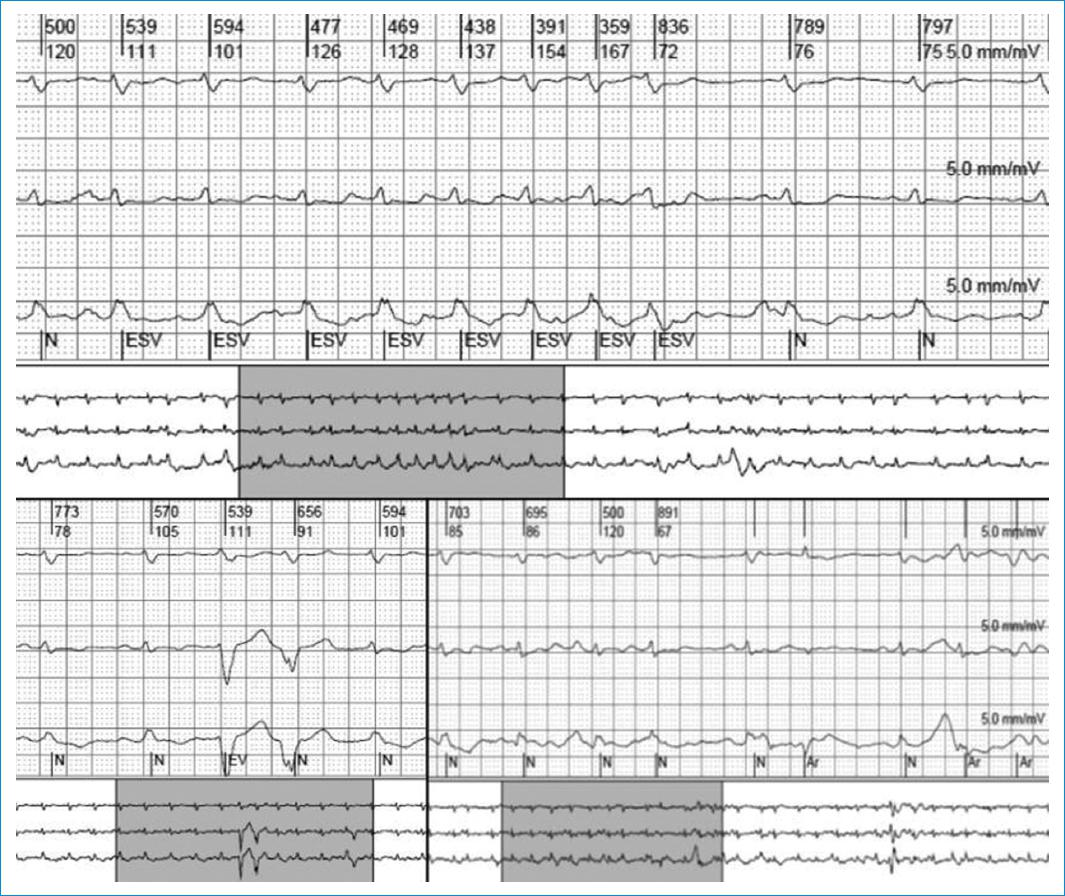
Paciente femenino de 67 años de edad, en seguimiento por hematología por anemia de cinco años de evolución, a quien se le realiza punción de médula ósea en la que se evidencian células plasmáticas con fenotipo alterado (> 10%) y concluye en el diagnóstico de mieloma múltiple. La analítica general y específica se resume en tabla 1. Evoluciona con clínica tórpida, desarrollando signos de IC por lo que se solicita electrocardiograma (Fig. 1), ecocardiograma Doppler color (Fig. 2) y resonancia magnética nuclear cardiaca. Esta última informa: ventrículo izquierdo no dilatado, leve incremento de espesor parietal septal (12 mm), fracción de eyección de ventrículo izquierdo: 51%. IM leve, AI moderadamente dilatada. Fibrosis difusa transmural inferoseptal basal e inferoseptal medial, fibrosis subendocárdica en pared inferior de ventrículo derecho. Valores de T1 mapping nativo aumentados en forma difusa (valores mayores a 1110 ms). Volumen extracelular (VEC) 55%, hallazgos compatibles con AC. Por referir palpitaciones se solicita estudio holter de 24 horas (Fig. 3). Se diagnostica mieloma múltiple y AC con IC función sistólica preservada, iniciando tratamiento dirigido a ambas enfermedades. En su seguimiento presenta mejoría clínica de la función renal y cardiaca; luego de dos meses de tratamiento, las mediciones de NT-pro BNP y troponina I ultrasensible habían descendido un 30%. En ecocardiograma control se puede objetivar mejoría de la función diastólica, progresando desde una disfunción diastólica III (flujo transmitral tipo restrictivo) a una tipo I (flujo transmitral prolongado), mejoría en Doppler tisular y parámetros de deformación longitudinal global.
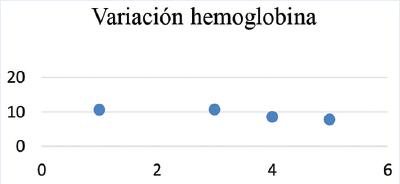
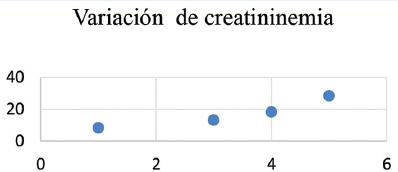
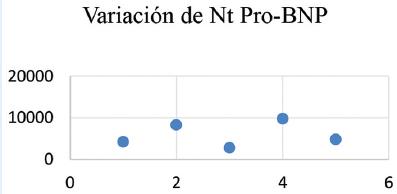
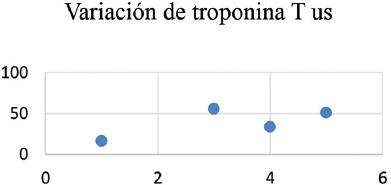
Tabla 1 Analítica general y tabla de dispersión de que muestra la evolución de hemoglobina, creatininemia, troponina T y NT Pro-BNP a lo largo del tiempo
| Prueba realizada | Resultado | Unidades | Valores de referencia | ||||
|---|---|---|---|---|---|---|---|
| Al momento del diagnóstico | Primer mes | Tercer mes | Sexto mes | Déci mosexto mes | |||
| Hematocrito | 34 | % | 37-47 | ||||
| Hemoglobina | 10.6 | 10,7 | 8,6 | 7,8 | g/dl | 12-16 | |
| Uremia | 42 | 107 | 210 | mg/dl | Menor a 71 | ||
| Creatininemia | 8.3 | 13,1 | 18,3 | 28,4 | mg/dl | 0.5-0.9 | |
| Na | 138 | mEq/l | 136-145 | ||||
| K | 4.2 | mEq/l | 3.5-5.1 | ||||
| Cl | 103 | mEq/l | 96-107 | ||||
| Clearance de creatinina | 59 | 50.4 | 34 | ml/min | Mujeres: de 88 a 128 ml/min | ||
| Proteínas urinarias | 925, 6 | 163.2 | mg/ 24 HS | Menor a 80 mg | |||
| Cadenas kappa | 16.1 | mg/l | 3.3 a 19.4 | ||||
| Cadenas lambda | 67.8 | 5.71 a 26.3 | |||||
| Cociente kappa/lambda | 0.23 | 0,26-1,65 | |||||
| Tronoponina T | 16.6 | 6 | 34 | 51 | Pg/l | 0-14 | |
| NT-Pro BNP | 4220 | 8259 | 2764 | 9746 | 4780 | pg/ml | Menor a 125 |
| Chagas | Negativo | ||||||
|
|
||||||
|
|
||||||
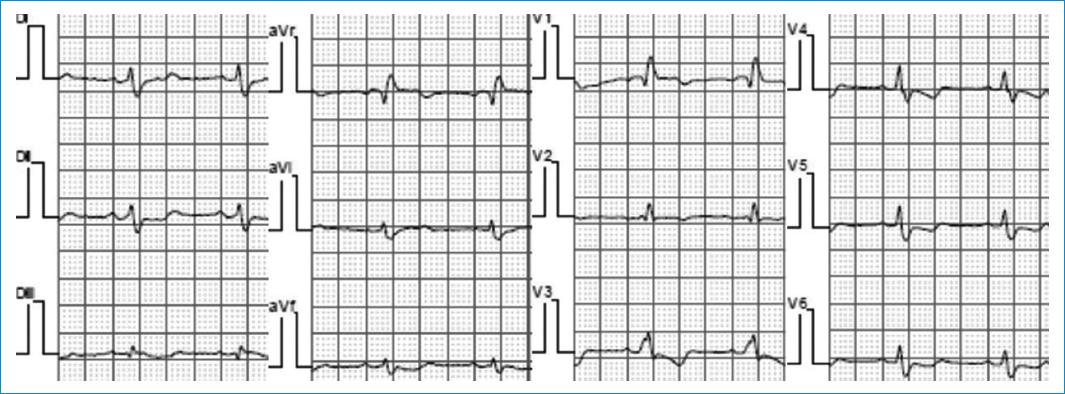
Figura 1 Electrocardiograma. Ritmo sinusal. Bajo voltaje. Bloqueo completo de rama derecha. Trastorno difuso de la repolarización. Q inferior sin criterios de fibrosis.
Epidemiología
Se trata de una enfermedad poco frecuente, que aumenta con la edad y en ciertos subgrupos de pacientes. Datos recientes informan una prevalencia de 40.5 casos por millón de habitantes y una incidencia de 14 casos por millón de personas por año. La población de pacientes mayores de 65 años con síndrome nefrótico presenta una incidencia mayor a la de la población general. En cuanto a la prevalencia, hay resultados divergentes; sin embargo, datos más recientes comunican que la amiloidosis AL sería un poco menos frecuente que aquella por transtiretina. Entre los casos de amiloidosis AL, aquella producida por cadenas lambda es más frecuente. Afecta más a hombres que a mujeres y la media de edad ronda los 56 a 63 años2.
Fisiopatología
Las células plasmáticas residen principalmente en la médula ósea y producen una gran variedad de anticuerpos, los cuales están compuestos por cadenas pesadas y livianas. Cuando un grupo de estas células toma un comportamiento maligno y comienza una replicación clonal, a su vez desencadena una producción excesiva de anticuerpos y las cadenas asociadas a dicho anticuerpo. A continuación, se presentan tres posibles escenarios:
– Que el clon de estas células solo tome una pequeña porción de la médula y la producción de cadenas livianas (CL) se excrete inofensivamente por la orina, lo que se denomina gammapatía monoclonal de significado incierto (que sucede en el 90% de estos casos).
– Que la invasión en la médula sea mayor y de lugar a anemia, lesiones líticas y, eventualmente, disfunción renal, lo que se denomina mieloma.
– En forma adicional a esta última situación, puede suceder que las cadenas livianas tomen un plegamiento anómalo y se depositen en los tejidos. Esta última condición se llama amiloidosis AL.
La disfunción cardiaca puede deberse a una proteotoxicidad secundaria a las cadenas livianas o los depósitos amieloides. También puede verse alteración de la membrana celular o toxicidad celular por el crecimiento de fibrillas, y formación de oligómeros de cadenas livianas solubles. Además, las CL pueden inducir apoptosis directamente, a diferencia de las fibrillas, que no lo hacen, e inducir la señalización de MAPK, lo que da como resultado una mayor producción de especies reactivas de oxígeno, alteración de la homeostasis del calcio, disfunción celular y, finalmente, muerte celular en cardiomiocitos adultos aislados3.
Los depósitos de amiloide AL se fijan en los tejidos y pueden provocar su disfunción y manifestaciones clínicas. Los órganos afectados con mayor frecuencia son los riñones (74%, que se manifiestan como albuminuria e insuficiencia renal), el corazón (60% típicamente con clínica de IC con función sistólica preservada), el tracto gastrointestinal (10-20% macroglosia, disfagia, estreñimiento, malabsorción, hemorragia digestiva), el hígado (27%, dando aumento de enzimas hepáticas e insuficiencia hepática en estadios avanzados) y el sistema nervioso autónomo (18%, presentando disfunción autonómica periférica). En el momento del diagnóstico, el 69% de los pacientes tiene más de un órgano afectado.
Diagnóstico
Presentación clínica
La presentación clínica dependerá principalmente del mayor órgano afectado. Los principales síntomas son astenia y disnea, que debido a su pobre especificidad muchas veces retrasan el diagnóstico.
Al momento del diagnóstico, dos tercios de los pacientes presentarán afectación renal, habitualmente con proteinuria en rango nefrótico4.
A diferencia de la amiloidosis ATTR, la AL se caracteriza por presentar sutiles cambios estructurales en el contexto de una florida y progresiva clínica de IC. Cerca del 50% de los pacientes tendrán afección cardiaca, manifestándose con síntomas de IC, astenia o intolerancia al ejercicio, que pueden ser acompañados por hipotensión, lo cual dificulta su manejo. Además, pueden presentar síncope, habitualmente en ejercicio, por restricción en el llenado, hipotensión y neuropatía. El compromiso cardíaco es el principal factor pronóstico ya que la causa de muerte se debe en un 75% a arritmias o IC avanzada, falleciendo alrededor de los 5 meses desde el inicio de los síntomas de IC si no se trata o a los 6 años si recibe tratamiento. Las arritmias supraventriculares son frecuentes y mal toleradas por su disfunción diastólica. Pueden afectar las arterias coronarias dando lugar a síntomas compatibles con síndrome coronario agudo.
El 20% de los pacientes mostrará enfermedad nerviosa periférica, como una polineuropatía sensitivo motora progresiva lenta, que habitualmente se manifiesta con dolor. Comúnmente presentan síndrome del túnel carpiano. La neuropatía autonómica es una complicación grave, que se observa con síntomas gastrointestinales (gastroparesia, diarrea, constipación, sangrado oculto, malabsorción, oclusión intestinal) aunque el 80% de las veces la invasión gastrointestinal es asintomática. Impotencia sexual e hipotensión ortostática son otras formas de manifestación.
Se diagnosticará macroglosia en un 15% de los pacientes. La afectación hepática suele verse como hepatomegalia y aumento de la fosfatasa alcalina sin signos de insuficiencia hepática. Pueden verse algunos signos de hipoesplenismo como cuerpos de Howell-Jolly junto a trombocitopenia. La expresión clínica de la afectación pulmonar resultará en insuficiencia respiratoria o nódulos en la forma localizada.
Cuando aparezca la expresión articular será una poliartropatía progresiva simétrica y bilateral, que en el caso de afectar la articulación del hombro aparece el signo patognomónico de la hombrera. La afectación muscular usualmente se asocia a compromiso cardiaco concomitante, y da el aspecto de persona atlética.
También pueden verse afectadas glándulas endocrinas dando como resultado síndrome de Sjögren, o insuficiencia adrenal o tiroidea. También se ha evidenciado en amiloidosis AL un mayor riesgo de sangrado, ya sea por la afectación vascular, déficit de factores de coagulación o aumento de proteínas fibrinolíticas5.
En la tabla 2 se resumen los principales signos y síntomas.
Tabla 2 Resumen de los principales signos y síntomas
| Sistema evaluado | Forma de manifestación habitual |
|---|---|
| Principales síntomas | Astenia y disnea de esfuerzo |
| Cardiovascular | Astenia o disnea de esfuerzo, intolerancia al ejercicio, hipotensión arterial, síncope Eventualmente con la evolución de la enfermedad también desarrollará signos de congestión pulmonar y sistémica |
| Renal | Proteinuria, síndrome nefrótico |
| Nervioso | Polineuropatía sensitivo-motora progresiva lenta, neuropatía autonómica, síndrome del túnel carpiano, estenosis espinal |
| Digestivo | Macroglosia, hepatomegalia, hipoesplenismo |
| Respiratorio | Insuficiencia respiratoria, nódulos pulmonares |
| Osteoarticular | Poliartropatía progresiva simétrica y bilateral |
| Sistema endocrino | Síndrome de Sjögren o insuficiencia adrenal o tiroidea |
| Hemostasia y coagulación | Fragilidad vascular, déficit de factores de coagulación o aumento de proteínas fibrinolíticas (sangrados espontáneos, equimosis) |
Exámenes complementarios
El diagnóstico de amiloidosis AL debe iniciarse solicitando cadenas ligeras libres de suero (kappa y lambda) y electroforesis de proteínas séricas y urinarias con inmunofijación. Como es sabido, la presencia de cadenas ligeras por sí sola no es específica para amiloidosis AL porque 20% de los pacientes con ATTR tienen una gammapatía monoclonal de significado incierto.
En la tabla 3 se presentan las principales recomendaciones en el tamizaje diagnóstico de amiloidosis AL.
Tabla 3 Recomendaciones generales para el manejo de un paciente bajo sospecha de amiloidosis
| Recomendaciones |
| –Confirmación en el tejido mediante biopsia y tinción con rojo Congo con la característica birrefringencia verde bajo luz polarizada. |
| –Confirmación mediante microscopía electrónica en el tejido de biopsia. |
| –Tipificación de la proteína mediante espectrometría de masa. |
| –Tipificación de la proteína mediante inmunomicroscopía óptica y/o electrónica, en la medida que haya anticuerpos confiables. |
| –Medición de las cadenas livianas libres séricas para evaluación de un trastorno proliferativo de células plasmáticas monoclonales. |
| –Inmunofijación sérica y urinaria para la evaluación de un trastorno proliferativo de células plasmáticas monoclonales. |
| –Medición de las cadenas livianas libres séricas, más la inmunofijación sérica y urinaria para la evaluación de un trastorno proliferativo de células plasmáticas monoclonales. |
| –Demostración de un trastorno proliferativo de células plasmáticas monoclonales mediante la demostración de plasmocitos clonales por la técnica más sensible disponible en la médula ósea para el diagnóstico de amiloidosis de tipo AL. |
Exámenes de laboratorio. Histopatología
El diagnóstico definitivo será la detección directa de material amiloide a través de un test de rojo Congo positivo de tejido afectado, preferentemente en forma no invasiva de tejido graso abdominal, recto o glándulas salivales, y de ser necesario (alta sospecha y resultado negativo) el órgano que clínicamente muestre compromiso. Las partes que sean positivas para rojo Congo deben correlacionarse con inmunohistoquímica o realizarse inmunofluorescencia directa, siendo preferible esta última por mayor tasa de éxito. En los casos que las pruebas rutinarias de inmunofluorescencia directa y/o inmunohistoquímicas no pueden tipificar definitivamente los depósitos de amiloide, la microdisección láser y el análisis proteómico basado en espectrometría de masas es una herramienta precisa y útil.
Biomarcadores
La Clínica Mayo recomienda de manera estandarizada dosificar la porción amino-terminal de péptido natriurético tipo B (NT pro BNP) y troponina ultrasensible (TnT us) y en 2012 incorporó la presencia de cadenas libres.
La combinación de NT pro BNP, TnT us y función renal también es útil en su estratificación. A su vez, la combinación de TnT us y dos parámetros derivados del strain cardiaco permiten una exactitud diagnóstica de aproximadamente el 98% sobre afección cardiaca en pacientes con amiloidosis AL6,7.
Otros biomarcadores novedosos (orientados a la fibrosis cardiaca), como la porción soluble de STS2 (sSTS2) y la galactina 3 pueden ser utilizados, pero solo el sSTS2 se comporta como marcador independiente de supervivencia en la amiloidosis Al8.
Electrocardiograma
La afectación del sistema de conducción es más común en la amiloidosis por transtiretina. Cambios característicos son el bajo voltaje frontal, especialmente si en otros exámenes como en el ecocardiograma, hay signos de hipertrofia y ondas Q en precordiales (aunque también pueden verse en derivaciones del plano axial) con un patrón conocido como pseudoinfarto o pseudofibrosis, el cual tiene implicaciones negativas en el pronóstico señalando menor sobrevida a un año y mayor impacto estructural9. Pueden observarse ectopias supraventriculares y ventriculares, siendo la arritmia más frecuente la fibrilación auricular y el flutter auricular (20%), habitualmente con trastornos de conducción (PR prolongado, bloqueo AV completo en 3%, trastornos de conducción intraventricular inespecíficos)10.
Holter y cambios electrofisiológicos
Puede observarse disminución de la variabilidad por disfunción autonómica, y arritmias de todo tipo. Tanto el nódulo sinusal como auriculoventricular suelen estar preservados, los tiempos infrahisianos usualmente son prolongados, mayores a 55 ms, lo cual es un predictor de muerte súbita11,12.
Ecocardiograma
Lo característico de esta enfermedad es que el infiltrado afecta aurículas y tabique interauricular, aumento simétrico del espesor parietal ventricular derecho e izquierdo con un aspecto brillante moteado tipo granular y sistema de conducción. Los cambios para comenzar a considerar el diagnóstico diferencial de AC son la relación Ee´ > 9.6, el volumen auricular izquierdo indexado y la disminución en la fracción de contractilidad miocárdica o fracción de acortamiento. Además, entre otros parámetros habituales pueden citarse el índice de excentricidad o espesor parietal relativo, ―ya que el fenotipo habitual en los pacientes con AC es de hipertrofia ventricular izquierda (HVI) concéntrica―, y el engrosamiento difuso valvar13.
Inicialmente, disminuye la complacencia auricular y aumenta la presión intraauricular, lo que lleva a que se ocupe menor tiempo tanto en el llenado ventricular como en el auricular. El descenso del llenado diastólico precoz es compensado por un enérgico llenado diastólico final, lo que, en estadios avanzados de la enfermedad, se traduce en una pseudonormalizacion del patrón. Por otro lado, otros autores han observado que la progresión de la enfermedad conlleva deterioro precoz de la diástole, pero no puede diferenciarse si este cambio se debe al envejecimiento normal celular. Es decir, que pueden observarse un patrón transmitral tipo pseudonormal, más habitual, o un patrón prolongado, el cual deberá diferenciarse de los cambios producidos por el envejecimiento. Otro aspecto a considerar es la afectación de la válvula mitral, ya que se ha observado una alta prevalencia de insuficiencia mitral que altera el patrón de llenado ventricular. Conforme avanza la enfermedad también se observan cambios en el patrón de las venas pulmonares. La evaluación de la función diastólica se correlaciona con la gravedad de la enfermedad y puede ser utilizada en su seguimiento14,15.
Resonancia magnética (RM)
Permite distinguir el fenotipo de miocardiopatía hipertrófica concéntrica, ya que se sabe que la AC tipo AL suele presentarse con forma simétrica, no así su homóloga por transtirretina (TTR)22. En la AC, el espacio extracelular expande su presencia con base en el depósito amiloide, lo que conduce a mayor concentración de gadolinio y, por tanto, a mayor realce tardío (RTG) en forma subendocárdica difusa o transmural16.
El mapeo en T1 antes de la administración de contraste (llamado T1 nativo) puede utilizarse para medir la señal intrínseca del miocardio, que, emparejado con el estudio posterior a la administración de gadolinio, puede utilizarse para calcular el VEC. Tanto el T1 nativo como el VEC están aumentados en amiloidosis AL o por TTR, incluso el T1 nativo parece estar aumentado antes que la evidente HVI, la RTG o los biomarcadores, y puede utilizarse en pacientes con deterioro importante de la función renal. Por otra parte, T1 es mayor en pacientes con amiloidosis AL, mientras que en su homólogo por TTR el VEC es preponderante17.
También es útil una señal de alta intensidad en T2, influenciada por el edema tisular. La señal en T2 se encuentra incrementada tanto en amiloidosis AL como por TTR, siendo más relevante en la primera, lo que da una idea de su fisiopatología18.
Gammagrafía ósea con radionucleótidos
El aporte principal es para la amiloidosis por TTR, ya que posee un alto valor predictivo positivo para ésta, y es menos útil con resultados intermedios.
Puede utilizarse el centellograma óseo para cuantificar la captación cardiaca del radiotrazador (habitualmente Tc-pirofosfato, TC-DPC, Tc hidroxymethylene DP). La evaluación se realiza en forma semicuantitativa, comparando la intensidad cardiaca con las costillas (método de Perugini) o con la región corazón/pulmón contralateral.
Tomografía por emisión de positrones
Se trata de una herramienta emergente para el diagnóstico de AC. Ciertos trazadores tienen afinidad por las proteínas plegadas tipo Beta e identifican el depósito amiloide independientemente del precursor, además de que tienen la ventaja de ser métodos cuantitativos19.
En la tabla 4 se presentan los estudios complementarios básicos a solicitar según el orden de complejidad, a sabiendas de que estos pueden ser útiles en el diagnóstico y el pronóstico de la enfermedad. En la tabla 5 se resumen los principales hallazgos de los estudios complementarios orientativos hacia la AC. En la figura 4 20 se presenta un algoritmo diagnóstico simplificado propuesto por el Grupo de trabajo de enfermedades del miocardio y pericardio de la Sociedad Europea de Cardiología. Del análisis se desprende que, de existir alta sospecha de este subtipo de enfermedad, la RM aporta más información que la gammagrafía y, acorde con la disponibilidad de estudios complementarios, este algoritmo podría simplificarse sin la utilización de la misma.
Tabla 4 Resumen de los principales estudios complementarios
| Histopatología | Rojo Congo positivo de tejido afectado, inmunohistoquímica o inmunofluorescencia, estudio genético, espectrometría de masas |
| Laboratorio específico | Medición cuantitativa de cadenas ligeras libres de inmunoglobulina en suero, detección cualitativa de cadenas ligeras monoclonales en suero u orina utilizando electroforesis de inmunofijación y biopsia de médula ósea con citometría de flujo o inmunohistoquímica |
| Laboratorio y estudios complementarios generales | Riñón: análisis de orina en 24 h, creatininemia, nitrógeno ureico; Corazón: ECG, ecocardiograma, RMN, gammagrafía, TnT us, NT pro-BNP; hígado: hepatograma; pulmón: Rx o tomografía de tórax, pruebas de función pulmonar; GI: albuminemia, hemoglobina (anemia), biopsia |
GI: gastrointnestinal; ECG: electrocardiograma; RMN: resonancia magnética nuclear; TnT us: troponina T ultrasensible; NT Pro BNP: porción amino terminal del péptido natriurético cerebral; Rx: radiografía.
Tabla 5 Principales aportes específicos y orientativos de los estudios complementarios habituales en el diagnóstico de amiloidosis cardiaca
| Estudios complementarios | Hallazgo sugestivo de amiloidosis | |
|---|---|---|
| Electrocardiograma | Bajo voltaje, patrón pseudoinfarto o pseudofibrosis, fibrilación auricular, bloqueo aurículo-ventricular de alto grado | |
| Ecocardiograma | Cambios estructurales | Hipertrofia ventricular con espesor mayor a 12 mm sin causa que lo justifique, cavidad pequeña, distribución simétrica. Aspecto granular. Espesor parietal relativo aumentado, usualmente mayor a 60 |
| Ventrículo derecho | Espesor aumentado (mayor a 5 mm), función sistólica disminuida (TAPSE <19) | |
| Aurículas | Engrosamiento tabique interauricular, ambas aurículas y su dilatación | |
| Función diastólica | Disfunción grado 2 o mayor. Relación E/e×aumentada (usualmente mayor a 11). La Velocidad de propagación suele ser normal (por ventrículo pequeño) | |
| Velocidad Doppler tisular | Disminuidas, menores a 8 o incluso 5 cm, relación diástole temprana/tardía (e×/a×) disminuye con la progresión de la enfermedad | |
| Función sistólica | Normal a leve disminución (afectación en estadios avanzados) | |
| Deformación longitudinal global | Afectación basal con relativa conservación apical (< -15). Relación strain longitudinal ápex/base > 2.9 | |
| Resonancia magnética nuclear | Fenotipo | Hipertrófico, simétrico o asimétrico. |
| Sin utilización de contraste | Mapeo en T1 nativo; señal en T2, marcador de edema tisular | |
| Con utilización de contraste | Determinación del VEC, distribución del realce tardío | |
| Centellograma óseo | Comparación con estructuras óseas aledañas por método de Perugini, escala del 0 al 3 Comparación con la región contralateral | |
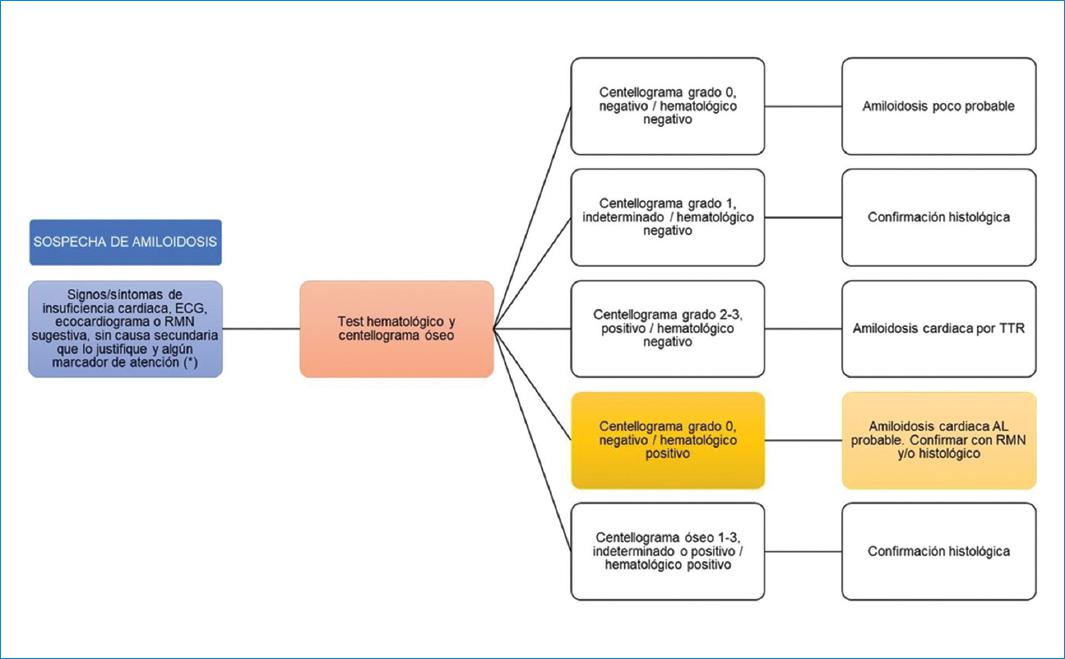
Figura 4 Algoritmo diagnóstico simplificado. (Adaptada de Garcia-Pavia P et al.20. *Los marcadores de atención o banderas rojas de amiloidosis son signos de alta sospecha de la enfermedad como paciente mayor de 65 años con insuficiencia cardiaca y fracción de eyección preservada, disfunción autonómica o sensitivo-motora, proteinuria o insuficiencia renal sin causa aparente, hematomas, strain con predominio de afectación basal, ECG con bajo voltaje, pseudofibrosis, trastornos de la conducción AV. ECG: electrocardiograma. RM: resonancia magnética.
Evaluación pronóstica
Para la evaluación estandarizada de pacientes con amiloidosis AL, la Clínica Mayo propone la utilización de TnT, NT-proBNP y técnicas de imagen21,22. Como se mencionó previamente, T1 nativo y la intensidad en T2 también aportan información pronóstica útil en este subtipo de la enfermedad.
Seguimiento
Si bien el NT-proBNP y los datos aportados por la ecocardiografía son los más utilizados en la actualidad, los mismos no identifican o cuantifican directamente la proteína amiloidogénica, sino sus consecuencias. Por lo tanto, la RM pareciera ser la técnica ideal, aunque contrasta por su menor disponibilidad.
Tratamiento
El tratamiento puede orientarse hacia la enfermedad de base o a sus diversas consecuencias en los sistemas implicados. Se hará hincapié en el sistema cardiovascular ya que presenta notables diferencias respecto de otras causas de IC. Por su impacto sistémico, esta enfermedad suele acompañarse de insuficiencia renal, síndrome nefrótico, hipotensión y disfunción autonómica, lo cual complica el manejo de la IC. Debe realizarse un seguimiento cercano del medio interno, función renal y clínica de los pacientes, indicarse una dieta reducida en sodio y pesarse diariamente para hacer seguimiento domiciliario de la volemia.
La utilización de inhibidores de la enzima convertidora de angiotensina o antagonistas del receptor de angiotensina suele ser mal tolerada y empeora los síntomas, y si bien el estudio PARAGON demostró beneficio en ciertos subgrupos para la utilización de sacubitrilo/valsartán, no se recomienda su uso generalizado.
Existe controversia respecto a la indicación de CDI ya que se sabe que no modifica el curso de la enfermedad, pero esto no ha sido evaluado en estudios aleatorizados en AC tipo AL; sin embargo, en vista de los últimos avances, existen subgrupos que podrían beneficiarse y se propone un sistema subcutáneo para disminuir el umbral de descarga23.
El uso de betablqueadores (BB) es inseguro debido a la tendencia a hipotensión y neuropatía de estos pacientes; además, el gasto cardiaco relativamente fijo depende de la taquicardia como mecanismo de compensación, Por consiguiente, de ser necesario su uso, se recomienda en la dosis más baja posible.
Los calcioantagonistas cronotrópico-negativos (verapamilo y diltiazem) suelen ser poco tolerados por su efecto inotrópico negativo intrínseco y vasodilatador, y, además, se ha observado afinidad por las proteínas amiloides lo que prolonga y determina incertidumbre en sus efectos adversos24,25. Tanto las frecuencias cardiacas bajas (menos de 60-80 latidos por minuto) como elevadas (mayores a 110-120) son mal toleradas.
Respecto al uso de digoxina es preferible en lugar de los BB en el contexto de un manejo agudo de fibrilación auricular de alta respuesta ventricular manteniendo dosis baja, tanto de carga como mantenimiento (< 0,8 ng/dl) y monitorizar frecuentemente la función renal, los electrolitos y la digoxinemia. Si bien se piensa que mantener el ritmo sinusal y preservar la fisiología normal trae beneficios en el contexto de la fisiología restrictiva, esto no ha sido comprobado en estudios a largo plazo26. Además, la contribución auricular en el llenado restrictivo es menor y existe una alta tasa de recidiva en el caso de reversión a ritmo sinusal. Como antiarrítmico debe pensarse en amiodarona y, en el caso de no poder establecerse un control eficaz, la ablación del nódulo AV e implantación de marcapaso puede ser una opción27.
Estos pacientes tienen un riesgo trombótico mayor, con la formación de trombos en la orejuela izquierda incluso en ritmo sinusal, por lo que en presencia de arritmias auriculares deben ser anticoagulados independientemente de su nivel de CHA2DS2-VASc u otros parámetros, como la velocidad de vaciado de la orejuela izquierda (que si bien se relaciona con aumento de eventos tromboembólicos, no debe guiar el tratamiento), y en el caso de fibrilación auricular en la cual se decida control del ritmo, debe hacerse previamente un ecocardiograma transesofágico. Por otro lado, debe cotejarse su riesgo de sangrado aumentado por mayor fragilidad capilar (incluso intestinal) o deficiencia de factor X, que incluso pueden contraindicar su anticoagulación.
Respecto al tipo de anticoagulante, existe evidencia tanto del uso de antivitamina K y anticoagulantes directos con similar eficacia y riesgos de sangrado, por lo que la decisión deberá ser tomada en forma interdisciplinaria28-30. En pacientes con disfunción autonómica grave e hipotensión sintomática puede utilizarse midodrina, iniciando 2,5 mg 3 veces por día, hasta una dosis de 10 mg, 3 veces por día31.
Una piedra angular en el tratamiento consiste en el mantenimiento de la euvolemia. El derrame pleural recidivante suele ser una constante, por lo que debe plantearse pleurocentesis, pleurodesis y colocación de tubos de drenaje en caso de complicación aguda o que la magnitud del mismo empeore los síntomas. Los edemas en las extremidades inferiores resultan beneficiados de la utilización de medias de compresión. Cuando estos son recidivantes o ante síntomas de congestión, deben utilizarse diuréticos del asa, solos o combinados con otro tipo de diurético (en este sentido, los antagonistas del receptor aldosterónico tienen beneficio clínico comprobado devenido del estudio TOPCAT). Sin embargo, estos fármacos pueden producir disminución del gasto cardiaco.
Pueden presentarse insuficiencia renal, síndrome nefrótico e incluso llegar a la necesidad de diálisis sanguínea o peritoneal (estas con similar supervivencia). En algunos casos puede indicarse transplante renal, cuando la remisión hematológica haya estado presente por al menos un año.
Debido a su habitual presencia de trastornos de la conducción auriculoventricular (AV), muchos pacientes presentan indicaciones clásicas para colocación de marcapaso o, en otras ocasiones, si se presentase bradicardia sintomática por neuropatía autonómica, en cualquier caso siempre se preferirá su modalidad bicameral para mantener la sincronía AV y la contribución auricular al llenado del ventrículo restrictivo. El trasplante cardiaco no es considerado una estrategia frecuente, debido al impacto sistémico de la enfermedad con otros órganos afectados, además del corazón trasplantado, lo cual aumenta la mortalidad en la espera del órgano y la mortalidad posterior.
Recientemente se han introducido los inhibidores del cotransportador sodio/glucosa para el manejo de la enfermedad y tienen relevancia tanto en el contexto de fracción de eyección reducida como preservada, independientemente de la presencia de diabetes mellitus tipo 2. A la luz de recientes ensayos clínicos y estudios sobre su mecanismo de acción, tienen beneficios particulares en la enfermedad, como no producir hipovolemia, bradicardia ni hipotensión significativas, aunque a la fecha se carece de estudios aleatorizados específicos en dicha enfermedad32-34.
Conclusiones
La AC tipo AL es una enfermedad poco frecuente sobre la cual están dándose importantes avances tanto diagnósticos como terapéuticos. Su identificación precoz traerá beneficio al paciente ya que el tratamiento radica principalmente en conocer la enfermedad de base y sostén de la IC, que constituye el principal factor pronóstico. El diagnóstico se basa en la identificación en sangre y orina de cadenas livianas, apoyado de su tipificación e imágenes cardiacas concordantes, y raras veces es necesaria la biopsia en la actualidad. La gammagrafía ósea es un método que aporta información para descartar su homóloga por transtirretina.

