











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
La hipertensión pulmonar es un síndrome que resulta de la restricción del flujo en la circulación pulmonar con un aumento > 20 mmHg en la presión media de la arteria pulmonar (PAPm) en reposo1,2.
Se han descrito múltiples vías patogénicas relacionadas con el desarrollo de hipertensión pulmonar. La hipertensión arterial pulmonar (HAP) caracteriza a un grupo de pacientes con hipertensión pulmonar precapilar (presión de cuña pulmonar ―PCP― < 15 mmHg y una resistencia vascular periférica (RVP) > 3 unidades Wood ―UW― en ausencia de otras causas de hipertensión pulmonar precapilar)1,2. La clasificación de los pacientes con hipertensión pulmonar permite su categorización en cinco entidades clínicas de acuerdo con su presentación clínica, hallazgos patológicos, características hemodinámicas y estrategias de tratamiento (Tabla 1)3.
Tabla 1 Clasificación clínica
| Grupo 1 | Hipertensión pulmonar arterial | Idiopática | |
| Familiar | |||
| Asociada a HAP | Trastornos del tejido conectivo | ||
| Cortocircuitos congénitos sistémicos a pulmonar | |||
| Hipertensión portal | |||
| Infección por VIH | |||
| Drogas y toxinas | |||
| Otras | |||
| Asociada a compromiso venoso o capilar | Enfermedad veno-oclusiva pulmonar | ||
| Hemangiomas capilares pulmonares | |||
| Grupo 2 | Hipertensión pulmonar con enfermedad cardiaca izquierda | Enfermedad auricular o ventricular izquierda | |
| Enfermedad valvular izquierda | |||
| Grupo 3 | Hipertensión pulmonar asociada con enfermedad pulmonar o hipoxemia | Enfermedad pulmonar obstructiva crónica | |
| Enfermedad pulmonar intersticial | |||
| Trastornos del sueño (respiratorios) | |||
| Desórdenes con hipoventilación alveolar | |||
| Exposición crónica a altas alturas | |||
| Anomalías del desarrollo | |||
| Grupo 4 | Hipertensión pulmonar por enfermedad trombótica o embólica crónica | Obstrucción por tromboembolia de las arterias pulmonares proximales | |
| Obstrucción por tromboembolia de las arterias pulmonares distales | |||
| Embolia pulmonar no tromboembólica (tumores, parásitos, objetos extraños) | |||
| Grupo 5 | Misceláneo | Sarcoidosis, histiocitiosis X, linfangiomatosis, compresión de vasos pulmonares (adenopatías, tumores, mediastinitis fibrosante) | |
HAP: hipertensión arterial pulmonar, VIH: virus de inmunodeficiencia humana.
Adaptada de: Patel MR et al.4
La prevalencia real de la HAP no está completamente identificada; reportes en la literatura muestran una prevalencia de 15/1.000.000 en Francia, mientras que en el Reino Unido es de 97/1.000.000; en los pacientes del grupo 1, se reportan prevalencias de 2.4 y 15-60/1.000.0002, y es una enfermedad más prevalente en mujeres, con una razón de 1.82. En Colombia, datos poblacionales estiman una prevalencia de 52.5/1.000.000, con una preponderancia femenina4.
La sospecha diagnóstica de la enfermedad se basa en síntomas y evaluación física, mientras el diagnóstico se realiza con la evaluación clínica, biomarcadores, pruebas de función cardiaca, ejercicio y función pulmonar, entre otros2. Lo anterior permite una evaluación integral del paciente. Adicionalmente, los parámetros clínicos, imagenológicos y hemodinámicos permiten valorar la gravedad de la enfermedad y realizar una estratificación de riesgo durante el proceso diagnóstico inicial y en las visitas de seguimiento. Existen múltiples escalas de estratificación de riesgo (Tabla 2), y es elemental la valoración de cambios en los parámetros hemodinámicos, funcionales y sintomáticos, y en la adherencia al tratamiento durante los seguimientos.
Tabla 2 Escala de estratificación del riesgo
| REVEAL6-8 | ESC/ERS2 | COMPERA9 | Registro francés10 | |
|---|---|---|---|---|
| Variables requeridas (n) | 12-14 | 9 | 8 | 4 |
| Pacientes basales (n) | 2.716 | 1.588 | 1.017 | |
| Asociación de HAP incluida | SI | Si | Si | No |
| Definición de bajo riesgo | < 6 puntos | 9 criterios de bajo riesgo | < 1.5 en puntaje promedio | 3-4 de 4 criterios de bajo riesgo |
| Mortalidad a un año por grupo (%) (bajo/intermedio/alto) | < 2.6 / 7 / > 10.7 | < 5 / 5-10 / > 10 | 2.8/9.9/21.2 | 1/NA/13-30 |
HAP: hipertensión arterial pulmonar; NA: no aplica.
Adaptada de: Galiè N et al.29
La HAP es una condición con una tasa de mortalidad importante que varía de acuerdo con el grado de estratificación del paciente (Tabla 2); aunque existen otras variables que influyen en la enfermedad (edad, género, comorbilidades), su impacto en el pronóstico no está esclarecido. Dada la asociación del riesgo con la mortalidad a un año, el objetivo del tratamiento es lograr que los pacientes estén en bajo riesgo asociado a una buena tolerancia al ejercicio, calidad de vida, función del ventrículo derecho (VD) y mortalidad2. En la vida real, la condición de bajo riesgo alcanza un porcentaje aproximado del 58% en el tercer año de diagnóstico, con lo cual persiste una importante población de riesgo intermedio que, durante la evolución de la enfermedad, puede llegar a ser del 48 y del 38%, en el primer y segundo año, respectivamente. En caso de persistir la estratificación de riesgo, su riesgo de muerte y necesidad de trasplante pulmonar es del 43% al tercer año de seguimiento5. Investigaciones recientes muestran el papel que desempeña el ventrículo derecho, así como la importancia del acople del ventrículo arterial, como herramientas adicionales en la discriminación de riesgo intermedio, con lo cual se ha planteado el posible beneficio de subdividir esta población en dos subgrupos: uno con comportamiento similar al riesgo bajo y otro hacia el riesgo alto. Bajo esta subdivisión del riesgo intermedio, se debe establecer la posibilidad de enfoques terapéuticos diferentes6.
Con la llegada de nuevas moléculas y estrategias de manejo, el tratamiento para la HAP ha evolucionado y mejorado su eficacia. Las intervenciones están compuestas de un abordaje inicial en el cual se toman medidas generales (actividad física, rehabilitación, soporte psicológico, prevención primaria y secundaria, terapias de soporte, estrategias para determinar la adherencia y referencias en caso de ser necesarias)2. La terapia específica con medicamentos se basa usualmente en el algoritmo de manejo de las guías de la Sociedad Europea de Cardiología y la European Respiratory Society ESC/ERS; estas incluyen bloqueadores de calcio, antagonistas de endotelina (ERA), inhibidores de fosfodiesterasa (PDE5i), estimuladores de guanilato ciclasa sGC estimulador (soluble), análogos de prostaciclinas y agonistas del receptor prostaciclina (IP)2. El tratamiento de la HAP se basa en cinco familias de medicamentos, que actúan por cuatro vías de señalización; estos pueden ser administrados como monoterapia, biterapia, terapia triple o terapia de transición. En este sentido, la terapia de transición permite la combinación de terapias con diferentes vías específicas para la enfermedad, y busca potenciar por medio de un sinergismo la terapia farmacológica5-9. Actualmente, existen posibles combinaciones entre las moléculas, con una contraindicación para la combinación de PDE5I y sGC estimulador10.
Justificación para un consenso
Las guías europeas (ESC/ERS) de 2015 sobre el diagnóstico y tratamiento de la HAP incluyen una extensa revisión sobre el manejo y diagnóstico de pacientes con hipertensión pulmonar2; sin embargo, existe un subgrupo de pacientes (aquellos del grupo 1 con riesgo intermedio) en quienes la definición de riesgo, a pesar de ser clara, agrupa pacientes no homogéneos en los cuales un análisis individualizado permite identificar factores de riesgo y diferencias en relación con la mortalidad. La estrategia de manejo debería ser diferente, esto relacionado con la inclusión de nuevas terapias farmacológicas y estrategias de tratamiento, las cuales permitirían un tratamiento más específico en función de los hallazgos particulares de cada paciente.
En Colombia se dispone de todas las opciones farmacológicas requeridas para el tratamiento de la HAP y, para los médicos involucrados en el manejo de la enfermedad es importante proveer herramientas ajustadas a nuestra realidad, para dar el espacio adecuado a cada opción terapéutica y saber qué puede aportar cada uno en términos de resultados.
Objetivos
Los objetivos del consenso realizado en el 2021 fueron:
− Discutir el manejo y la clasificación de pacientes adultos con hipertensión pulmonar del grupo 1 con riesgo intermedio, de acuerdo con los parámetros de las guías ESC/ERS 2015.
− Resaltar recomendaciones clínicas de manejo y sus limitaciones.
− Proveer recomendaciones clínicas basadas en la evidencia y la experticia para pacientes con hipertensión pulmonar del grupo 1 con riesgo intermedio.
− Adaptar estas recomendaciones al contexto colombiano, de acuerdo con la disponibilidad, la accesibilidad y los recursos del sistema de salud.
− Definir los vacíos existentes en el manejo de pacientes con hipertensión pulmonar del grupo 1 con riesgo intermedio.
− Discutir posibles estrategias de manejo y líneas de posible investigación a futuro.
Método
Se invitaron 15 expertos (cardiólogos y neumólogos) de diferentes áreas de medicina y que tienen contacto en pacientes con hipertensión pulmonar, así como experticia en el manejo de esta enfermedad. De estos, la totalidad (15 /15) aceptaron participar en el proceso de la elaboración del consenso.
Se realizó un conjunto de preguntas propuestas por el moderador del consenso y basadas en la literatura científica disponible (n = 14). Estas preguntas fueron socializadas con el grupo de expertos para su complemento y discusión. Con base en estas, se realizó una búsqueda sistemática de la literatura para cada una de ellas.
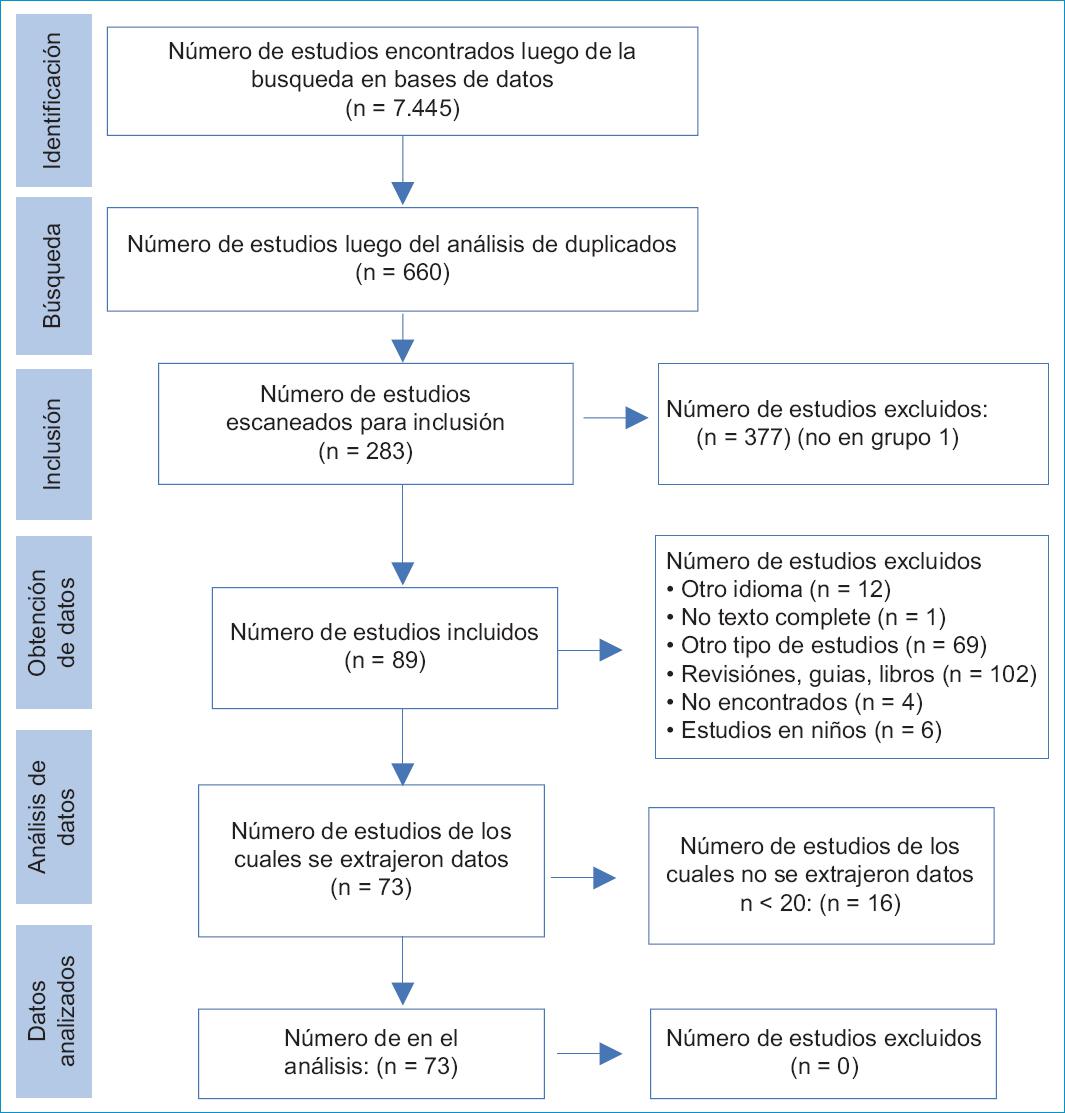
Se realizó una búsqueda de acuerdo con las guías PRISMA para elaboración de revisiones sistemáticas. Esta se hizo en BVS, Cochrane Library, PubMed, Medline y Web of Science; con los criterios MeSh y DeCs sin límite de idioma o tiempo. Se encontraron 7.445 artículos, de los cuales se incluyeron 73, distribuidos en cuatro grupos generales:
− Estratificación y manejo (n = 30)
− Tratamiento (n = 25)
Cuatro subgrupos por molécula
− Selexipag (n = 2)
− Iloprost (n = 1)
− Riociguat (n = 5)
− Treprostinil (n = 6)
− Costo-eficacia (n = 3)
− Consideraciones especiales (n = 1)
Se extrajeron los datos por tres operadores independientes en un formato de Microsoft Excel® (versión 16.54). Dos investigadores de cada artículo incluido realizaron una valoración metodológica por medio del sistema de GRADE. Las dudas frente a inclusión y valoración metodológica fueron resueltas por un tercer investigador. Esta información fue compartida con los expertos para su interpretación y lectura.
Posterior al análisis de la literatura por los expertos, se desarrolló una serie de encuestas en línea en el formato de encuesta Google Forms®; el proceso para las respuestas de las preguntas permitió una respuesta abierta para obtener la mayor información posible. Se realizó una primera votación que buscó determinar las respuestas en formato de opción múltiple para las preguntas. Esto se logró por medio de una votación sobre las preguntas con las que se ejecutó la búsqueda (en formato abierto) y las subsecuentes respuestas (en formato opción múltiple), que derivaron de la información encontrada en la literatura. Se estableció un punto de corte del 70% de favorabilidad para la inclusión de la respuesta (resultados en suplemento 1). En una segunda votación, se solicitó que votaran a un total de 13 recomendaciones (suplemento 2) en una escala tipo Likert del 1-9; siendo 1 no pertinente y 9 muy pertinente. Adicionalmente, se brindó la oportunidad de proveer un texto libre para avalar su respuesta, complementar la recomendación o aportar información adicional. La valoración de los resultados de esta fase se basó en la metodología Delphi modificada (o RAND); se determinó consenso cuando la recomendación alcanzó un punto de corte del 80%. Los textos libres fueron evaluados por los investigadores y se usaron como complemento de la discusión sobre la recomendación. Con los resultados de esta votación, se realizó una reunión virtual en la que se discutieron los resultados de las preguntas sin consenso; durante esta reunión, se tomó la decisión de expandir seis recomendaciones sin consenso que abarcaban múltiples temas, en un total de diecisiete nuevas preguntas abiertas con sus respectivas respuestas en opción múltiple. Sobre estas preguntas se siguió la metodología propuesta en el primer momento de las votaciones y una vez se estructuraron las respuestas, se plantearon once nuevas recomendaciones que fueron sometidas a votación con la metodología Delphi modificada propuesta en la segunda fase de votación (suplemento 3). Las variables cualitativas de la primera votación se expresaron en porcentajes, mientras que las variables de la segunda votación se expresaron en mediana y rango intercuartil (IQR).
Resultados y discusión
Se reportó consenso en dieciocho de veinte preguntas (90%) (Suplemento 2 y 4). Las recomendaciones y su respectivo sustento en la evidencia y discusión se describen a continuación.
− Se debe realizar la estratificación de riesgo en pacientes con hipertensión arterial pulmonar del grupo 1 con riesgo intermedio, con los parámetros descritos en la figura 2 (de mayor a menor relevancia clínica).
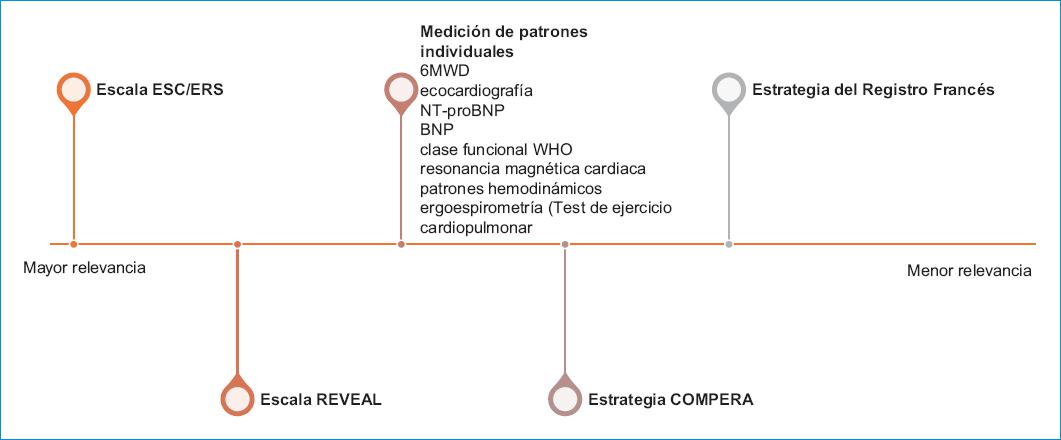
Mediana: 9 (IQR 1.25)
A la fecha, en la literatura médica no hay una única estrategia que aporte información robusta de pronóstico y diagnóstico en pacientes con HAP. Es vital la estratificación en el primer año de diagnóstico, dado que el estado de riesgo en este año parece ser crítico en predecir el estado de riesgo que alcanzará el paciente a largo plazo11,12. Las guías clínicas de manejo y consensos de especialistas sugieren la determinación de deterioro clínico, progresión o enfermedades concomitantes, la función del VD, así como su estabilidad o suficiencia, el estado actual del paciente y su pronóstico a largo plazo2. Lo anterior, es más factible por medio de un abordaje multidimensional que idealmente permita medir capacidad de ejercicio y aporte información del VD2.
A pesar de esto, los expertos recomiendan, en primer orden de relevancia, la estrategia REVEAL, dado que dentro de la revisión sistemática realizada es la que cuenta con la mayor evidencia para su uso13-18, así como capacidad de discriminar eficientemente pacientes en riesgo, como se demuestra en una cohorte norteamericana de 1.632 pacientes donde el modelo de riesgo REVEAL 2.0, discriminó el riesgo tanto en pacientes con diagnóstico previo de la enfermedad, como en pacientes con HAP incidente14. Adicionalmente, la versión REVEAL Lite 2 ha demostrado ser un método robusto, de fácil aplicación, que permite la estratificación a pesar de ser una versión reducida de la escala REVEAL19. Es importante esclarecer que un estudio, en particular de 140 pacientes, reporta como limitante de la escala REVEAL la estratificación de pacientes con riesgo intermedio20. La escala ESC/ERS también es una estrategia exitosa, que permite la discriminación de riesgo y, por ende, predecir la supervivencia; es sensible al cambio y permite determinar que aquellos pacientes que cambian de riesgo modifican su supervivencia a largo plazo21.
En caso de tener múltiples mediciones ya sea por escala o individuales, se deben ponderar estos resultados y considerar, de acuerdo con el peor resultado, la optimización del manejo. Las estrategias complementarias permiten aumentar el poder predictivo, lo cual cobra una mayor importancia en un grupo heterogéneo, como son los pacientes con riesgo intermedio; estrategias de mayor costo, como la resonancia magnética cardiaca o la ergoespirometría deben ser consideradas en casos seleccionados. El panel de expertos es consciente de que no todos los centros y puntos de atención del país cuentan con la infraestructura para realizar todos los parámetros de rutina (ejemplo, pruebas cardiopulmonares o pruebas de ejercicio); en estos casos, puede usarse la medición de parámetros individuales y en la literatura se cuenta con guías que permiten determinar el manejo acorde con las medidas individuales21-24. Por su lado, el registro COMPERA calcula el promedio individual de riesgo al asignar un puntaje de 1-3 (1= bajo riesgo, 2= riesgo intermedio, 3= alto riesgo) y promediando con la media de cada variable. En contraste, el registro francés cuenta el número de variables con valores de bajo riesgo24; a pesar de estas diferencias, el resultado tiende a ser similar, especialmente en el seguimiento a un año. Sin embargo, el registro francés parece ser más preciso en determinar pacientes con buena sobrevida a largo plazo en comparación con el COMPERA para evaluar el riesgo24.
Lo anterior hace pertinente, a futuro, la realización de ensayos clínicos o estudios científicos robustos, en los que sea posible la comparación de las escalas, las estrategias y los parámetros individuales, su sensibilidad y especificidad en términos de mortalidad y morbilidad y que se logre una estratificación robusta de pacientes con hipertensión pulmonar, especialmente en aquellos con riesgo intermedio en quienes es más difícil la estratificación y su correlación con la supervivencia a largo plazo.
− La estratificación en pacientes del grupo 1 con hipertensión pulmonar se debe iniciar desde el momento del diagnóstico, con revaloración de la estratificación dada la sensibilidad al cambio que tienen las escalas.
Mediana: 9 (IQR 1)
Esta recomendación se basa en que es un hecho establecido que determinar el riesgo dentro del primer año de tratamiento, permite tipificar a los pacientes y estratificar riesgos a largo plazo11. Esta conducta está recomendada por las guías de manejo2,12,25,26.
Por otra parte, la evaluación de riesgo durante el seguimiento del paciente identifica subgrupos con diferentes respuestas al tratamiento, y, por consiguiente, diferentes pronósticos a largo plazo11,15,17; entre estos, son particularmente relevantes aquellos pacientes que logran una disminución en el riesgo, que directamente se asocia con una respuesta adecuada al tratamiento y es uno de sus objetivos2.
En el contexto colombiano es importante aclarar que, al igual que la estratificación, los seguimientos van sujetos a la disponibilidad de cada centro y que, dadas las barreras en la atención en salud, es importante la estratificación en cada consulta por medicina especializada, de modo que permita optimizar el enfoque y tratamiento de los pacientes.
− La estratificación en pacientes con hipertensión arterial pulmonar del grupo 1 con riesgo intermedio ayuda a determinar el riesgo de morbilidad y mortalidad.
Mediana: 9 (IQR 1)
Esta recomendación se fundamenta en estudios que reportan cómo la estratificación de los pacientes permite determinar la supervivencia a un año20,27y, en algunos estudios prospectivos, hasta cinco años28; así mismo, como se mencionó previamente, permite valorar la respuesta a la terapia y realizar los ajustes necesarios. Un análisis posthoc del subestudio SERAPHIN mostró una reducción en la morbilidad y mortalidad en los pacientes que alcanzan el objetivo de bajo riesgo29.
− La subclasificación de riesgo intermedio, en riesgo intermedio bajo y riesgo intermedio alto, aporta utilidad clínica a los pacientes con hipertensión arterial pulmonar del grupo 1 y se debe realizar de acuerdo con la gravedad de la disfunción del VD y el número de hospitalizaciones de los pacientes con HAP del grupo 1.
Mediana: 8 (IQR 1.25)
La recomendación se sustenta en que los pacientes que pertenecen al grupo intermedio han demostrado ser un conjunto heterogéneo, y que tal vez no sea posible estratificarlos en un solo grupo. En el seguimiento a largo plazo, se evidencia que algunos logran las metas terapéuticas de disminuir a bajo riesgo, mientras otros permanecen en riesgo intermedio, sin lograr modificar su supervivencia a largo plazo o progresan a alto riesgo. Esto supone la posibilidad de que dentro del grupo intermedio se puedan observar dos subgrupos de pacientes (alto y bajo riesgo intermedio). Un estudio en 301 pacientes permitió demostrar diferencias en la supervivencia a largo plazo en este subgrupo de pacientes, evidenciando adicionalmente, que tal vez el mejor parámetro para discriminar este subgrupo es la función del ventrículo derecho30.
A la fecha, existe poca evidencia que permita hacer una fenotipificación adecuada de los dos subgrupos de pacientes dentro del grupo de pacientes con riesgo intermedio, así como de la medida más adecuada para la estratificación de estos. El grupo de expertos considera que el VD, al ser un determinante de síntomas y sobrevida31, podría ser un protagonista central en la clasificación y por esto se plantea su uso como parte fundamental de la clasificación. A futuro, sería interesante contar con estudios que permitan tipificar estos subgrupos, además de definir el parámetro principal de medición para determinar disfunción ventricular derecha.
− En pacientes con hipertensión pulmonar del grupo 1 con riesgo intermedio, es posible clasificar a los pacientes en dos subgrupos de riesgo de acuerdo con los hallazgos de la resonancia magnética (RM) cardiaca, el número de hospitalizaciones y los parámetros ecocardiográficos. Se recomienda que uno de los parámetros ecocardiográficos de mayor importancia para esta clasificación es la evaluación de la función del VD. Cabe aclarar que esta recomendación es una orientación clínica dado que, a la fecha, hay suficiente evidencia sobre la heterogeneidad de los pacientes en riesgo intermedio, mas no de su clasificación en dos subgrupos.
Mediana: 8.5 (IQR 1)
Como se mencionó previamente, la evidencia es poca para estratificar estos dos subgrupos de pacientes; sin embargo, a la luz de demostrar que unos pacientes son respondedores a terapia (logran tener riesgo bajo) y otros no, dentro del grupo intermedio, los expertos consideran recomendar que los parámetros para valorar la función ventricular derecha (RM cardiaca y ecocardiograma) y la respuesta al tratamiento (número de hospitalizaciones) se planteen como una estrategia de valoración de estos pacientes. No obstante, a la fecha no están claros los puntos de corte, las variables específicas o el paraclínico ideal para discriminar estos dos subgrupos. Se plantea que la RM cardiaca o la ecocardiografía en tres dimensiones (3D) podrían ser las imágenes diagnósticas ideales; sin embargo, no hay suficiente evidencia en la literatura ni experticia que permita determinar por medio de una escala de riesgo los subgrupos.
− En pacientes con hipertensión pulmonar del grupo 1 con riesgo intermedio y que presentan disfunción grave del ventrículo derecho, puede ser considerada la adición de una prostaciclina parenteral.
Mediana: 9 (IQR 1)
Esta recomendación se fundamenta en ensayos clínicos que avalan la evidencia del uso de prostaciclinas parenterales y la mejoría de los parámetros de ejercicio32. Por otro lado, esta estrategia se encuentra recomendada por la ESC/ERS 2015, en pacientes con clase funcional, de acuerdo con la Organización Mundial de la Salud (World Health Organization, WHO-CF), III y IV (IIa/c)2, mientras que un consenso de expertos la recomienda para pacientes con WHO-CF III con progresión rápida de la enfermedad u otros marcadores de mal pronóstico, al igual que pacientes con WHO-CF IV33y el consenso canadiense lo recomienda en pacientes con escala de la New York Heart Association (NYHA) CF IV. Todos los anteriores son pacientes con parámetros de riesgo intermedio-alto que, al tener una clase funcional tan limitada, el compromiso del ventrículo derecho grave está explícito y, por ende, el tratamiento con estas moléculas es una opción farmacológica correcta.
− Es razonable adicionar un agonista del receptor IP en pacientes con hipertensión pulmonar del grupo 1 con riesgo intermedio y que presentan disfunción leve a moderada del VD.
Mediana: 8.5 (IQR 2.25)
Esta recomendación se basó en las guías de la ESC/ERS 2015, que aconsejan su uso en pacientes con WHO-CF II y WHO-CFIII (I/B). Lo anterior se sustenta en el ensayo fase 3 de la molécula, en el que se evidencia una disminución del 40% del riesgo de morbilidad/mortalidad2,8.
Cabe aclarar que esta recomendación se realiza para pacientes que presentan hospitalizaciones por falla cardiaca derecha o que persisten con parámetros hemodinámicos de riesgo intermedio [definidos como presión aurícula derecha (PAD) 8-1.4 mmHg, índice cardiaco (IC) 2-2.4 L/min/m2y/o SatvM 60-65%].
− Es razonable realizar terapia de transición intraclase en pacientes con hipertensión pulmonar del grupo 1 con riesgo intermedio y que presentan disfunción leve a moderada del VD.
Mediana: 7 (IQR 2.5)
Los expertos no encontraron consenso para esta recomendación. Es probable que esto se deba a que la transición en pacientes con hipertensión pulmonar es un concepto novedoso y nace de la necesidad de tener estrategias de tratamiento en aquellos cuya enfermedad progresa rápidamente o requieren manejos parenterales constantes. Esto, acompañado del advenimiento de nuevas moléculas, plantea una estrategia de intervención interesante. Estudios estiman que para 2008 aproximadamente el 10% de los pacientes habían intentado terapia de transición; sin embargo, la evidencia científica que apoye este conocimiento es escasa. Estudios posteriores que evalúan nuevas moléculas han demostrado la factibilidad de la transición, así como su potencial en pacientes con riesgo bajo e intermedio. Igualmente, estudios como REPLACE permiten demostrar el beneficio de este tipo de moléculas en poblaciones individualizadas con determinadas características34, datos que son acordes con la recomendación del WSPH 2018, que indica la terapia de transición como una medida benéfica que se debe acompañar de la experticia clínica35. Es importante enfatizar que esta recomendación no alcanzó el consenso de expertos, dado el bajo número de estudios que se tienen a la fecha (cabe recordar que es un concepto novedoso) al igual que lo mencionado en las guías ERS/ECR. Sin embargo, se considera que son necesarios estudios de mayor tamaño, más robustos y con un seguimiento posterior de los resultados; surgirá nueva evidencia que permita hacer una recomendación. De igual forma, se espera que en las guías ERS/ECR 2022 se cuente con una recomendación internacional para estas moléculas.
Lo que sí es claro es que se debe monitorizar a los pacientes durante y después de la transición y basarse en el perfil clínico y paraclínico individual de cada uno para realizar dicha transición.
− En pacientes con hipertensión pulmonar del grupo 1 con riesgo intermedio se recomienda evaluar su adherencia al tratamiento.
Mediana: 9 (IQR 0)
Para el panel de expertos, la evaluación de la adherencia consiste en una estrategia esencial que permite determinar si la falla terapéutica es realmente una falla o una pobre adherencia. La diferenciación de estas dos entidades permite establecer estrategias de tratamiento que tienden a ser más efectivas para el control de la enfermedad. Es importante que dentro de esta línea de recomendaciones quede claro que la falta de adherencia no es causal de escalonamiento terapéutico; por tanto, se debe indagar el porqué de la no adherencia y, cuando sea posible, buscar otras vías de administración si esta es la limitante para la adherencia, así como crear conciencia en los pacientes y educarlos frente a su enfermedad y los riesgos que derivan de no ser adherente al tratamiento. Lo anterior va de la mano de las guías ESC/ERS que recalcan la importancia de una evaluación periódica de adherencia dada la complejidad de la HAP y la posibilidad de cambios en el régimen terapéutico por los pacientes, sus familiares o médicos inexpertos en el tema2.
− Se recomienda la reevaluación con las herramientas de estratificación de los pacientes con hipertensión pulmonar del grupo 1 con riesgo intermedio, que no cumplen metas terapéuticas. El periodo de reevaluación debe estar acorde con el subgrupo de riesgo.
Mediana: 9 (IQR 1)
Para los expertos, la evaluación regular de todos los pacientes con hipertensión pulmonar es recomendada por las guías de manejo2. Se indica un abordaje multidimensional de acuerdo con la recomendación 1; el tiempo recomendado por las guías ESC/ERS es cada 3 a 6 meses si hay cambio de terapia, seguimientos parciales cada 3 a 6 meses (valoración física, laboratorios básicos, electrocardiograma y caminata de 6 minutos 6MWD) y valoraciones con todos los parámetros de las escalas cada 6 a 12 meses2. En aquellos pacientes que no cumplen metas terapéuticas con buena adherencia a medicamentos, es importante determinar la progresión de la enfermedad y descartar enfermedades concomitantes, clase funcional y pronóstico a largo plazo.
− Se define escalada terapéutica en pacientes con hipertensión arterial pulmonar del grupo 1 y con riesgo intermedio, como la adición tercera opción terapéutica a pacientes en terapia combinada doble de una vía de acción diferente.
Mediana: 9 (IQR 1)
La escalada terapéutica es una estrategia de tratamiento cada vez más frecuente para el manejo de pacientes con HAP2; es una estrategia válida que ha sido usada en otras enfermedades, y, a la fecha, se considera uno de los estándares de manejo para estos pacientes36. Su uso se encuentra fundamentado por ensayos clínicos y estudios observacionales5,7-9,37-40, revisiones sistemáticas41y guías de manejo2. Es una terapia efectiva, que permite una reducción en el deterioro clínico, aumenta la 6MWD y disminuye la resistencia vascular periférica, con una incidencia de eventos adversos similar entre grupos, sin un impacto significativo en la mortalidad2. De acuerdo con lo anterior, los expertos realizan esta recomendación.
− Se debe realizar escalonamiento terapéutico ascendente en pacientes con hipertensión pulmonar del grupo 1 con riesgo intermedio, cuando no hay cumplimiento de las metas terapéuticas, no se tolera la vía de administración, hay una mala adherencia al tratamiento y hay presencia de efectos secundarios.
Mediana: 9 (IQR 1)
En aquellos pacientes que no muestran cumplimiento de metas ante el escalonamiento ascendente de medicamentos, el concepto permite considerar la intervención simultánea combinando las tres vías de señalización diferentes que se encuentran involucradas en la enfermedad, definidas como la vía del óxido nítrico, prostaciclina y endotelina2. Adicionalmente, como se describe en la recomendación 13, esta estrategia de triple terapia ha mostrado efectividad y seguridad suficientes para recomendarla42.
El panel de expertos recomienda una evaluación minuciosa de aquellos pacientes con mala adherencia al tratamiento, intolerancia a la vía de administración o presencia de eventos secundarios, o ambas; no está recomendada la escalada terapéutica en estos pacientes, sino la evaluación de la terapia instaurada, la evaluación de la no adherencia o de la no tolerancia de vía, así como la determinación de eventos adversos (leves o graves); este último paso primordial al realizar escalada terapéutica pudiese incrementar el riesgo de eventos adversos. Sin embargo, el cambio de terapia en estos grupos de pacientes podría terminar en escalada terapéutica y por esto son incluidos en la recomendación.
− En pacientes con hipertensión pulmonar del grupo 1 con riesgo intermedio, que reciben tratamiento combinado (doble terapia), se recomienda uso de riociguat como estrategia de transición o triple terapia en pacientes con clase funcional de la OMS II-III, HAP idiopática o hereditaria, HAP relacionada con trastornos del tejido conectivo o HAP asociada a cardiopatías congénitas. Posterior al inicio de riociguat se espera encontrar mejoría de la 6MWD, cambio en la clase funcional, cambios en los niveles de NT-proBNP, cambios en la escala de Borg y retraso en el deterioro clínico de la enfermedad.
Mediana: 8 (IQR 2)
La evidencia para realizar esta recomendación se basa en un ensayo clínico previamente mencionado, en el que fue posible identificar en qué subpoblación de pacientes riociguat demuestra un beneficio luego del inicio del tratamiento43. Estudios posteriores de seguimientos en la vida real y en otros observacionales demuestran una mejoría significativa de la 6MWD, la RVP, los niveles de NT-proBNP y la disminución de la clase funcional6,7,34,44-51. En el seguimiento a largo plazo, estas mejoras se mantienen en el tiempo (1 año) con una tasa de sobrevida del 97%7,47, tanto en pacientes en monoterapia como en aquellos en combinación con ERA o prostanoides. Es importante recalcar que no es una estrategia para el manejo de HAP con disfunción sistólica grave y que la combinación de riociguat con PDE-5i se encuentra contraindicada2.
− El tratamiento con agonistas del receptor IP, como selexipag, está indicado en pacientes con hipertensión arterial del grupo 1 y riesgo intermedio, en pacientes entre los 18 a 75 años con diagnóstico de HAP relacionada con trastornos del tejido conectivo, asociada a cardiopatía congénita corregida e idiopática o hereditaria; pacientes que tengan una marcha de 6 minutos con valores entre 282-434 m y estén en terapia estable con ERA + inhibidor endotelina-1. En estos pacientes se espera encontrar, asociados al tratamiento, reducción en la morbimortalidad y en la tasa de hospitalización, retraso en el deterioro de la enfermedad, mejoría en la 6MWD y cambios en la clase funcional, con un retraso al tiempo de requerir prostanoide parenteral.
Mediana: 8.5 (IQR 1)
El uso de selexipag ha demostrado en ensayos clínicos una reducción de la RVP en la semana 1717. Los datos frente a la caracterización demográfica y las variables de desenlace derivan de este estudio17y muestran la subpoblación que mejor responde a la terapia con selexipag. En un ensayo clínico controlado robusto con importante número de pacientes (GRIPHON)8y otros estudios observacionales, se ha mostrado que su uso aditivo a monoterapia o terapia combinada se asoció con reducción de la morbilidad y mortalidad (40%), tasa de hospitalizaciones, deterioro clínico, trasplante pulmonar y cambio de terapia2,52,53. Por lo anterior, los expertos recomiendan el uso de selexipag en este grupo de pacientes y recomiendan realizar seguimiento con las variables descritas.
− La adición de iloprost puede ser considerada en pacientes con hipertensión pulmonar del grupo 1 y riesgo intermedio que se encuentran en biterapia que no toleren los otros manejos médicos o no acepten su uso. Sin embargo, es importante considerar que, para el panel de expertos, la baja adherencia podría ser un factor limitante para su uso.
Mediana: 8 (IQR 2.5)
Las guías de manejo (ESC/ERS) recomiendan el uso de iloprost inhalado (IB-IIb/C) y endovenoso (IIA/C y IIB/C)2,26,33. Resultados de ensayos clínicos muestran un aumento en la capacidad de ejercicio con mejoría sintomática y de la resistencia vascular periférica en pacientes con hipertensión pulmonar hereditaria o asociada al tejido conectivo39,54. Con lo anterior, los expertos recomiendan su uso; sin embargo, unas de las principales limitantes que evidencian los expertos en su práctica diaria y que no deben ser interpretadas como una contraindicación sino como una oportunidad para individualizar terapias, son la baja adherencia y la poca aceptabilidad por parte de los pacientes.
− Es razonable adicionar treprostinil en pacientes adultos (> 18 años) del género femenino con hipertensión pulmonar del grupo 1 y riesgo intermedio que se encuentran en biterapia y que presenten clase funcional III de la NYAH, HAP idiopática o hereditaria, HAP asociada al tejido conectivo, HAP asociada a cardiopatías congénitas y 6MWD de 163-547 m. Posterior al inicio de treprostinil se espera encontrar cambio en la clase funcional, mejoría de la sobrevida, mejoría de la 6MWD; cambios en el deterioro clínico, mejoría en el índice de disnea de Borg, mejoría en la RVP; mejoría en el índice cardiaco, mejoría en la PAPm; reducción en la tasa de hospitalización y menor requerimiento o uso de terapias adicionales.
Mediana: 8.5 (IQR 1)
La razón fundamental para esta recomendación deriva del ensayo clínico donde se evidencia la subpoblación de pacientes que mejor responde a cada medicamento55,56. Los expertos realizan la recomendación del uso de treprostinil basándose en que muestra una mejoría en la RVP y en la capacidad de ejercicio y disminuye el perfil de riesgo55-60.
Es importante discutir que la recomendación en género femenino no excluye a los pacientes de género masculino. Treprostinil, al igual que todas las terapias, puede ser usado en cualquier género; no obstante, la recomendación se basa en un ensayo clínico en el que predominó el género femenino55,56.
− No se cuenta con información suficiente que permita determinar el costo-eficacia de la escalada terapéutica entre tratamientos orales y parenterales.
Mediana: 7 (IQR 3)
Para los expertos, esta recomendación no alcanzó nivel de consenso, dado que, a pesar de encontrar tres estudios de fármaco-eficacia61-63, la información resulta insuficiente para poder realizar este análisis y, por ende, determinar esta recomendación.
− El nivel educativo, la condición cultural, el tipo de pagador, la ubicación geográfica; la presencia de comorbilidades, la polifarmacia y la vía de administración del medicamento son factores limitantes para la formulación y la adherencia a distintas terapias en HAP.
Mediana: 9 (IQR 0.25)
Esta recomendación se basa tanto en la experticia de los médicos del consenso, como en un estudio64que demuestra que los pacientes con estratos socioeconómicos más bajos tienen mayor riesgo de mortalidad y deterioro clínico, independiente de las características clínicas o factores de riesgo. Sin embargo, para el panel de expertos es importante resaltar que, a la fecha, faltan estudios que midan el acceso que tienen los pacientes con HAP en Colombia, en especial aquellos que viven lejos de las ciudades capitales.
Conclusiones
El análisis de la gravedad de los pacientes con HAP mediante el uso protocolizado de estrategias multiparamétricas es relevante, pertinente y vinculante para la toma de decisiones terapéuticas farmacológicas e incluso el trasplante pulmonar. La clasificación debe realizarse tanto en el momento del diagnóstico, como en las visitas de seguimiento. La terapia farmacológica no puede ser considerada homogénea pues la probabilidad de combinaciones posibles en una escala clínica de las más usuales puede alcanzar un sinnúmero de alternativas, utilizando las diferentes escalas de riesgo disponibles. Además, existen modificadores como las expectativas y la autonomía del paciente, empoderamiento en su tratamiento, aspectos relacionados con el pagador, grupos sociales y características culturales o de acceso a los centros asistenciales. Al día de hoy no se dispone de un argumento farmacogenómico para la toma de decisiones terapéuticas en términos de número de fármacos o secuencia ideal, pero la evidencia permite definir que dos fármacos son mejor que uno y tres son mejor que dos, en la búsqueda de la modificación del riesgo del paciente.
En este consenso se realizan una serie de recomendaciones sobre estratificación, tratamiento y otros apartados que implícitamente resaltan la necesidad de mayor número de estudios científicos de buena calidad, que permitan determinar el comportamiento de los apartados mencionados. Es necesario disponer de datos propios respecto a las características clínicas de los pacientes con HAP en nuestro entorno de país, acceso y disponibilidad de medicamentos, desenlaces, costos, y, en general, la integralidad de la ruta de atención con sus respectivos defectos y oportunidades de mejora. Es posible que acciones gremiales, como este consenso, aporten elementos de valor para optimizar la atención de los pacientes con HAP en Colombia.

