










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista Facultad de Ingeniería Universidad de Antioquia
versão impressa ISSN 0120-6230
Rev.fac.ing.univ. Antioquia no.77 Medellín out./dez. 2015
https://doi.org/10.17533/udea.redin.n77a04
ARTÍCULO ORIGINAL
DOI: 10.17533/udea.redin.n77a04
Aggregation study of asphaltenes from colombian Castilla crude oil using molecular simulation
Estudio de agregación de asfaltenos provenientes del petróleo crudo colombiano de campo Castilla utilizando simulación molecular
Jennifer de León-Barreneche1*, Bibian Alonso Hoyos-Madrigal1, Wilson Antonio Cañas-Marín2
1Facultad de Minas, Universidad Nacional de Colombia. Carrera 80 # 65-223 - Núcleo Robledo. A. A. 1027. Medellín, Colombia.
2Instituto Colombiano del Petróleo (ICP), ECOPETROL S.A. Kilómetro 7 vía Piedecuesta. A. A. 678. Piedecuesta, Colombia.
* Corresponding author: Jennifer de León Barreneche, e-mail: jde@unal.edu.co
(Received August 4, 2014; accepted May 11, 2015)
ABSTRACT
A molecular simulation model to study the mechanism for asphaltene aggregation is presented. Four species were selected, obtained from structural analysis of asphaltenes from the oil extracted from Castilla oil field, in Colombia. Energetic contributions to the aggregation process for each species were studied, and the solubility parameter was evaluated. Finally, the aggregation process between different species was studied in order to determine the tendency of the molecules towards self-association. Results show that for all species, aggregation state is energetically favorable; also, both Van der Waals interactions and electrostatic forces contribute equally to the aggregation process. It was also found that the molecular structure of the substances has a big influence on the manner in which asphaltenes aggregate. For continental structures, long ramifications cause a physical obstacle for aggregation. On the other hand, the molecular flexibility associated with archipelago structures enables the aggregation with other species, but somehow hinders the process of self-association. The solubility parameter for all four substances was within the range established by literature.
Keywords: Asphaltene, molecular simulation, aggregation energies, solubility parameter
RESUMEN
Se presenta un modelo de simulación molecular para estudiar el mecanismo de agregación de asfaltenos. Se seleccionaron cuatro especies, obtenidas a partir de análisis estructurales del petróleo extraído del campo Castilla, en Colombia. Se estudiaron las contribuciones energéticas al proceso de agregación y se calculó el parámetro de solubilidad para cada especie. Finalmente, se estudió el proceso de agregación entre las diferentes especies con el fin de determinar la tendencia de las moléculas hacia la autoasociación. Los resultados muestran que para todas las especies, el estado de agregación es energéticamente favorable, y que tanto las fuerzas de Van der Waals como las fuerzas electrostáticas contribuyen en igual magnitud al proceso de agregación. Se encontró también que la estructura molecular de las moléculas tiene una gran influencia en la manera cómo se agregan los asfaltenos. Para las estructuras continentales, las ramificaciones largas ocasionan un impedimento físico para la agregación. Por otro lado, la flexibilidad asociada a las moléculas tipo archipiélago favorece la agregación con otras especies, pero de alguna manera entorpece la autoasociación. El parámetro de solubilidad para todas las especies se encontró dentro del rango establecido por la literatura.
Palabras clave: Asfalteno, simulación molecular, energías de agregación, parámetro de solubilidad
1. Introduction
Asphaltene related problems are one of the most important causes of production loss in the petroleum industry. Asphaltenes, widely known as the ''cholesterol of petroleum'', have the tendency to form aggregates. Due to changes in pressure, temperature and composition, this aggregates precipitate from the crude oil and in times deposit, which can result in formation damage [1] . Formation damage can present in different ways. First, asphaltenes can form aggregates of sufficient size to reduce oil permeability and ultimately obstruct the flow of the mobile phase of hydrocarbons. Second, the asphaltene aggregates can deposit on mineral rock and alter its original wettability, thus reducing the mobility of the flowing phase. In addition, asphaltene aggregates can deposit on other surfaces, such as flowing pipes, security valves and other surface equipment, posing as a real threat to the safety and success of the oil operation.
Asphaltenes are a solubility class of crude oil, soluble in aromatic solvents like benzene or toluene, and insoluble in low boiling-point n-alkanes, such as n-pentane and n-heptane. Due to the tendency of asphaltenes to form aggregates, obtaining an accurate structure to represent all the features of asphaltenes has become a difficult task. In this sense, literature clearly defines two tendencies in asphaltene representation models: on the one hand, asphaltenes are often represented as a single structure [2, 3] , either derived from available experimental data [4] or developed from average features of asphaltene molecules, such as molecular weight and heteroatoms content [5] . From experimental data, general features of asphaltene molecules have been widely studied and established [5-7] . Asphaltenes are complex molecules, consisting on polyaromatic-condensed rings with aliphatic chains and some heteroatoms, most commonly sulfur, nitrogen and oxygen, and in some cases, small amounts of metals like vanadium, nickel and iron. Average molecular weight of asphaltenes range from 500 to 1000 daltons [7, 8] .
On the other hand, some researchers tend to agree that it is not accurate to represent asphaltenes as a single molecule, but rather as a set of molecules that reunite all the features of asphaltenes. In this regard, asphaltene systems have often been represented as a mixture of various (often three) structures with different sizes and characteristics, and that can be considered individually as asphaltene molecules [9-12] .
The model chosen to represent asphaltenes directly affects the way aggregation is studied. The use of a single molecule to represent the entire population of asphaltenes surely translates into more simplicity in the system, and thus less compute time. However, a single molecule fails to represent all the features of asphaltenes and still fall into the requirements an asphaltene molecule should have, such as molecular weight and heteroatom content of the molecule. The polydisperse model, although more accurate in describing the real nature of the system, implies more compute time and propose a technical difficulty in determining the actual forces that intervene in asphaltene aggregation. Recently developed theories are inclined to consider a supramolecular model for asphaltene aggregation that even include the content of heavy metals in the molecule structure, a feature often neglected due to the low fraction of these elements in the structural analysis of asphaltenes [13] . This level of detail allows depicting the association behavior of asphaltenes observed experimentally. However, large systems consisting of many asphaltenes plus solvent molecules might be difficult to simulate due to high computational cost.
The aim of this work is to understand the way in which asphaltene molecules form aggregates. Molecular dynamics is used to evaluate the solubility parameter and the aggregation mechanism of four different asphaltene molecules. The structures used herein were derived from previous experimental work conducted in oil extracted form Castilla oil field, in Colombia. Even though the controversy on the asphaltene model has not yet been solved, this study helps understanding the way in which these molecules interact with each other, and the forces that influence this interaction. The interaction energy is discretized in its different contributions. The interaction between each molecule and molecules of different nature is also evaluated.
2. Model
Figure 1 shows the molecular structure of the four asphaltene molecules used in this study, A1, A2, A3 and A4. These molecules were adapted from molecules obtained in previous studies as a result of experimental analyses conducted on colombian Castilla crude oil [14] . It can be seen from Figure 1 that the molecules A1, A2 and A3 (1a, 1b and 1c) have a continental structure, with a core made of condensed aromatic rings surrounded by saturated chains of hydrocarbons. The molecule A4, (1d), has an archipelago structure, with small groups of rings connected by saturated chains. Molecule A1 (Figure 1a) presents a core made of eight aromatic rings and one atom of sulfur and one atom of nitrogen. Molecule A2 (Figure 1b) has a similar structure, but has a larger core made of 15 aromatic rings, and one more atom of sulfur.
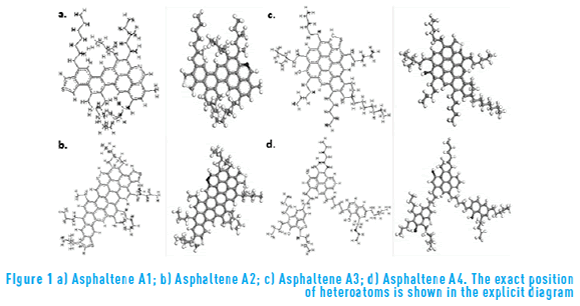
Molecule A3 (Figure 1c) has a condensed core consisting on nine aromatic rings, but has an oxygen and a sulfur atom, and no nitrogen atoms. This molecule also has longer saturated ramifications on its structure. Molecule A4 (Figure 1d) has three aromatic groups connected with saturated chains. It also has three oxygen atoms and one sulfur atom, and no nitrogen.
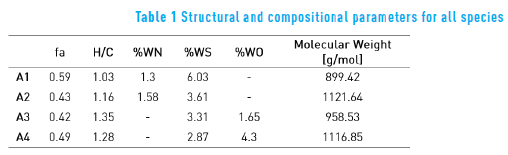
Table 1 shows the structural and compositional parameters for all four asphaltene molecules. The first column contains the aromatic carbon fraction (fa); the second column contains the hydrogen to carbon (H/C) ratio; the three following columns represent the heteroatoms content in weight percentage for nitrogen, sulfur and oxygen, respectively; finally, the last column contains the molecular weight of the species. For all species, these structural features are within range for an average asphaltene molecule [5] .
The molecules were represented in an atomistic framework, in which each atom is considered individually using the LAMMPS package and the consistent valence force field CVFF [15] . The interaction energy expression is shown in Eq. (1):
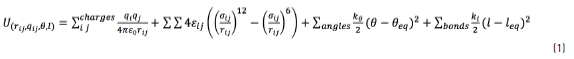
where ε and σ are the Lennard-Jones potential parameters, rijis the separation distance between atoms, qiand qjare the atomic charges; kθ , θ and θeq are the angle constant, the angle between each three atoms and the equilibrium angle, respectively; and kl , l and leq are the bond constant, the bond length between each pair of atoms and the equilibrium bond length, respectively.
Lennard-Jones parameters and bond and angle constants were assigned considering the nature of the atoms that form the pair, bond or angle, respectively [15] . Partial charge for each atom was assigned using the Equivalent Charge method [16] .
Once all the molecules were constructed and all the parameters were assigned, interaction energies and the solubility parameter were calculated. The solubility parameter was evaluated as stated in Eq. (2):
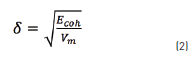
where Ecohis the asphaltene cohesion energy and Vm is the molar volume of each species. The cohesion energy of a substance can be defined as the energetic difference between a molecule in isolated conditions, emulating ideal gas state, and the molecule in the bulk of a phase, as a condensed state. The ideal gas state is simulated as the molecule being in a vacuum, with no neighbors. The condensed state is represented as the molecule surrounded by its replicas and placed in a simulation box with periodic boundary conditions in all three dimensions. The different contributions to non-bond energy in both states were evaluated, and the difference between them was determined.
3. Simulation details
This study was carried out in three stages. First, an energy minimization was conducted for all four molecules at 0 K in order to obtain the most adequate configuration for each molecule individually. Once the optimum structures were found, molecular dynamic simulations were performed in an NPT ensemble at a temperature of 300 K and a pressure of 200 atm for each molecule in a simulation box with reflective walls in all three dimensions. This simulation allows calculating the energy of each molecule in vacuum, emulating the ideal gas state.
Second, molecular dynamic simulations of twelve replicas of each molecule were conducted, using an NPT ensemble at the same temperature and pressure with periodic boundary conditions in all three dimensions. This simulation allows calculating the energy of each molecule while surrounded by its replicas, emulating the condensed state. This simulation also allows calculating the molar volume of each molecule.
Third, the interaction of each molecule with molecules of distinct nature was evaluated. To this purpose, the combinations A1A2, A1A3, A1A4, A2A3, A2A4 and A3A4 were placed in an NPT simulation box, at 300 K and 200 atm. The interaction energies were evaluated.
The cut radius for each species was selected as the length of the molecule, to guarantee adequate distance for interaction between the molecules. The Ewald's summation method was used to evaluate the long-range electrostatic interactions. The SHAKE algorithm was used to maintain bond length. A complete list of the parameters used in this simulation can be found elsewhere [17] .
Molecular dynamic simulations of at least 10 ns were conducted (9.5 ns equilibration and 0.5 ns production), using a time step of 1 fs. Non-bond energies (Van der Waals and electrostatic) were evaluated. Molar volume was estimated and the solubility parameter was calculated for all four species.
4. Results
Figure 2 shows the results for non-bond energy of molecule A1 in isolated conditions and Van der Waals, electrostatic and total non-bond energy for A1 surrounded by its replicas with periodic boundary conditions after an equilibration period of 9.5 ns. As can be seen from the figure, the average energy for the molecule in periodic conditions is lower than the average energy for the isolated molecule. That is, the molecule is more stable when it is in aggregated state. This is consistent with the association behavior observed in asphaltene molecules.
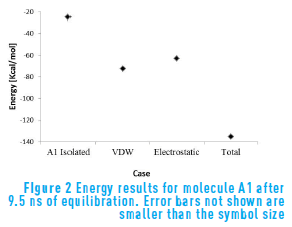
Regarding the forces that intervene in the aggregation process, Figure 2 shows that Van der Waals energy is the main force contributing to the formation of aggregates, even though the electrostatic contribution is in the same order of magnitude and cannot be neglected. The Van der Waals energy is associated with the aromatic rings in the core of the structure, while the electrostatic contribution is associated mainly with the content of heteroatoms.
Figure 3 shows the energetic results for species A2. Molecule A2 is similar to molecule A1, with a core composed of a greater number of aromatic rings and the presence of one more sulfur atom. This can be evidenced in the energy results, being the Van der Waals energy lower, and the electrostatic energy higher for molecule A2 compared with molecule A1. This indicates that the presence of point charges generates a lower contribution to aggregation for continental structures.
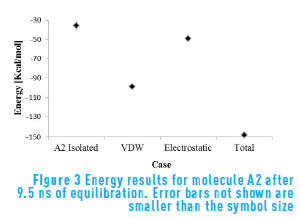
Figure 4 shows the energetic results for molecule A3. This molecule presents similar behavior than the previous cases, except that in this case the molecule has higher energy contributions, mostly due to a lower electrostatic contribution. The presence of an oxygen atom, which has a higher partial charge than the other heteroatoms, along with the rigid continental structure cause molecule A3 to have lower interaction energy than the other two molecules.
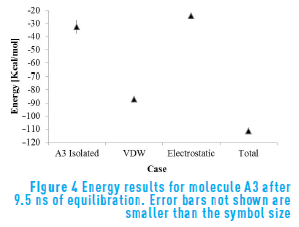
Figure 5 presents the energetic results for molecule A4. As expected, the energy results corroborate the association behavior observed experimentally. This molecule has a different, more flexible structure than the previous cases. That flexibility allows this structure to reorientate in space so it can balance the partial charges. Therefore, even though this molecule has three oxygen atoms in its structure, the energy results show that the interaction energy is higher and this molecule forms stable aggregates.
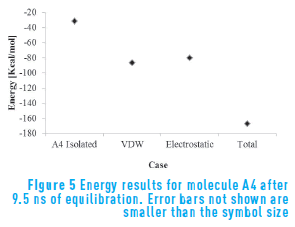
For molecule A4, the electrostatic and Van der Waals contribution are similar, and neither can be neglected. This means that the effect of the flexibility of the molecule combined with the partial charges associated with oxygen atoms have a combined effect on aggregation, originating more stable aggregates.
Table 2 summarizes the results obtained for molecular volume, the cohesive energy and the solubility parameter for all four molecules. The solubility parameter is characteristic for each pure substance, and gives an indication to the behavior of a substance when mixed with another. For instance, reported experimental value for toluene is 18.3 MPa1/2, and for benzene is 18.7 MPa1/2 [8] . This indicates that these two substances are able to mix with each other, while n-heptane, with a solubility parameter of 15.3 MPa1/2 [8] is incompatible with either of them.
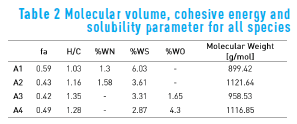
As asphaltene do not have a definite structure, literature reports a range of values for solubility parameter, between 17.0 and 21.0 MPa1/2 [8, 18, 19] . Results obtained in this study are within expected for average asphaltene molecules. In the case of molecule A3, the solubility parameter is slightly lower than expected. This is associated with the energy difference between the isolated and the condensed state of this molecule, which was lower than expected due to the presence of the oxygen atom in its structure.
Figure 6 shows the total interaction energy between molecule A1 and the other three molecules. It can be seen that the lowest energy state is achieved when molecule A1 interacts with molecule A4, which has the flexible structure. That is, the interaction with molecule A4 is stronger than for the other molecules (even itself), in spite of the high content of heteroatoms.
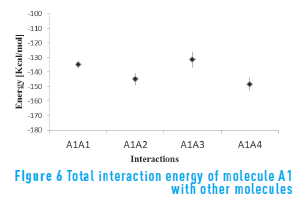
The energy of molecule A1 in presence of itself and molecule A2 is similar, and is lower than the energy of molecule A1 in presence of molecule A3. This suggests that the ramifications of molecule A3 hinder the aggregation process with other continental structures. Figure 7 depicts a snapshot of A1A3 interaction. From the figure, it is possible to observe how the long ramifications of molecule A3 are an obstacle to the aggregation of the two molecules.
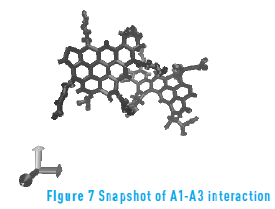
Figure 8 shows the energy of molecule A2 in presence of the other three molecules. For molecule A2 in presence of A1 it is possible to see a strong interaction, even stronger than with itself. This strong interaction energy could explain why, if they were in deed two separate molecules, the experimental procedure took them to be one.
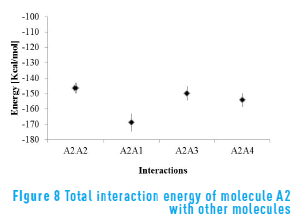
Figure 9 shows the energy results for molecules A3 in presence of other molecules. Molecule A3 has higher interaction energy with itself than with the other structures, both continental (A1 and A2) and archipelago (A4). This corroborates the assumption that the long saturated chains of this structure create a steric effect that hinders aggregation.
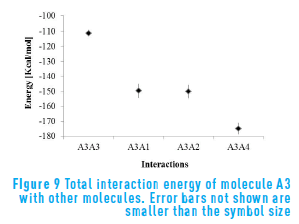
Figure 10 shows the interaction energy for molecule A4 in presence of other structures. Molecule A4 has the lowest interaction energy of all molecules, even though it is higher with itself than with the continental structures. This means that the flexible structure of molecule A4 forms more stable aggregates with the continental structures. Molecule A4 behaves in a similar manner to the presence of A1 and A2, and interacts with a little high energy in presence of molecule A3. In addition, in spite of the three oxygen atoms present in the structure of molecule A4, it is evident that the molecule A3 is more stable when it interacts with the flexible structure of this molecule.
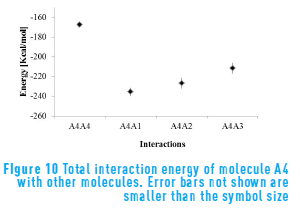
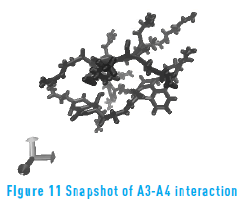
Figure 11 depicts a snapshot of the interaction between molecules A3 and A4. It is possible to observe form the figure how molecule A4 surround molecule A3 in order to maximize the interaction between the aromatic rings. This ability to rearrange its geometry is what gives molecule A4 the high values of interaction energy.
5. Conclusions
Results obtained in this work allow concluding that molecular dynamic models are adequate to represent asphaltene systems. The energy results are consistent with the association behavior of asphaltenes observed experimentally. For the asphaltenes considered here, Van der Waals and electrostatic contributions are equally relevant in governing the aggregation process. The Van der Waals energy associated with the aromatic content, and the electrostatic energy associated with the presence of heteroatoms, are strictly tied to the structural and compositional features of each molecule, thus for molecules with different structure and content of heteroatoms results may vary.
Although asphaltenes molecules show a tendency towards self-association, the interactions between continental and archipelago structures exhibit higher association energy. The presence of long ramifications in continental structures hinders the formation of asphaltene aggregates, while the flexibility of archipelago structures allows these molecules to balance the partial charges in its structure and enables the aggregation behavior.
6. References
1. S. Punnapala and F. Vargas, ''Revisiting the PC-SAFT characterization procedure for an improved asphaltene precipitation prediction'', Fuel, vol. 108, pp. 417-429, 2013. [ Links ]
2. J. Pacheco, I. Zaragoza and J. Martínez, ''Asphaltene Aggregation under Vacuum at Different Temperatures by Molecular Dynamics'', Energy & Fuels, vol. 17, pp. 1346-1355, 2003. [ Links ]
3. E. Buenrostro, C. Lira, A. Gil and J. Wu, ''Asphaltene precipitation in crude oils: Theory and experiments'', AIChE J., vol. 50, pp. 2552-2570, 2004. [ Links ]
4. E. Rogel, ''Simulation of Interactions in Asphaltene Aggregates'', Energy & Fuels, vol. 14, pp. 566-574, 2000. [ Links ]
5. L. Buch et al., ''Molecular size of asphaltene fractions obtained from residuum hydrotreatment'', Fuel, vol. 82, pp. 1075-1084, 2003. [ Links ]
6. O. Mullins, E. Sheu, A. Hammami and A. Marshall, Asphaltenes, Heavy Oils, and Petroleomics, 1st ed. New York, USA: Springer, 2007. [ Links ]
7. O. Mullins et al., ''Advances in Asphaltene Science and the Yen − Mullins Model'', Energy & Fuels, vol. 26, pp. 3986-4003, 2012. [ Links ]
8. L. Vicente, C. Soto, H. Pacheco, J. Hernández and J. Martinez, ''Application of molecular simulation to calculate miscibility of a model asphaltene molecule'', Fluid Phase Equilib., vol. 239, pp. 100-106, 2006. [ Links ]
9. T. Takanohashi, S. Sato, I. Saito and R. Tanaka, ''Molecular Dynamics Simulation of the Heat-Induced Relaxation of Asphaltene Aggregates'', Energy & Fuels, vol. 17, pp. 135-139, 2003. [ Links ]
10. T. Takanohashi, S. Sato and R. Tanaka, ''Structural Relaxation Behaviors of Three Different Asphaltenes Using MD Calculations'', Pet. Sci. Technol., vol. 22, pp. 901-914, 2004. [ Links ]
11. S. Stoyanov, S. Gusarov and A. Kovalenko, ''Multiscale modelling of asphaltene disaggregation'', Mol. Simul., vol. 34, pp. 953-960, 2008. [ Links ]
12. I. Mackie and G. DiLabio, ''Importance of the Inclusion of Dispersion in the Modeling of Asphaltene Dimers'', Energy & Fuels, vol. 24, pp. 6468-6475, 2010. [ Links ]
13. M. Gray, R. Tykwinski, J. Stryker and X. Tan, ''Supramolecular Assembly Model for Aggregation of Petroleum Asphaltenes'', Energy & Fuels, vol. 25, pp. 3125-3134, 2011. [ Links ]
14. L. Navarro, M. Álvarez, J. Grosso and U. Navarro, ''Separación y caracterización de resinas y asfaltenos provenientes del crudo castilla. Evaluación de su interacción molecular'', CT&F Ciencia, Tecnol. y Futur., vol. 2. pp. 53-67, 2004. [ Links ]
15. S. Plimpton, ''Fast Parallel Algorithms for Short – Range Molecular Dynamics'', J. Comput. Physics, vol. 117, pp. 1-19, 1995. [ Links ]
16. A. Rap and W. Goddard, ''Charge Equilibration for Molecular Dynamics Simulations'', J. Phys. Chem., vol. 95, pp. 3358-3363, 1991. [ Links ]
17. M. Levitt, M. Hirshberg, R. Sharon and V. Daggett, ''Potential energy function and parameters for simulations of the molecular dynamics of proteins and nucleic acids in solution'', Comput. Phys. Commun., vol. 91, pp. 215-231, 1995. [ Links ]
18. F. Frigerio and D. Molinari, ''A multiscale approach to the simulation of asphaltenes'', Comput. Theor. Chem., vol. 975, pp. 76-82, 2011. [ Links ]
19. A. Tharanivasan, ''Asphaltene Precipitation from Crude Oil Blends, Conventional Oils, and Oils with Emulsified Water'', Ph.D. dissertation, University of Calgary, Calgary, Canada, 2012. [ Links ]
