











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink1. Introduction
Low carbon steels (LCS) are the most widely used materials in different industries for the parts, equipment and structures manufacture, this due to their low cost with respect to alloyed steels. LCS has good mechanical strength and ductility, low hardness and good welding behavior. However, most applications of the elements manufactured with these steels are exposed in highly contaminating environments where metal corrosion is favored, which can lead to deterioration of mechanical properties and in more critical cases destruction and change of materials. This leads to increase costs for maintenance, repair and in many cases puts in risk of failure the installed infrastructure during the operation. For this reason, it has been of great interest to study the conditions and environments both external and internal to which the steel is exposed, in order to obtain information to prevent critical damages, to identify the effects or products of corrosion and to be able to predict behaviors for both short and long-term[1,2]. Among the sources of corrosion in which LCS are exposed the media containing sulfates appears[3]; chlorides, sulfur[1], salts, carbonates, particulate matter[4]. Also, chemical species associated with combustion products in boilers, storage of reagents and fluids transport in pipelines.
Corrosion in carbon steels is manifested by the surface oxide layer formation. This oxide layer has been of great interest in the advance investigations on the degradation of carbon steels[2-6]. It has been sought to identify and characterize the complex composition of the layer. This layer is generally composed of lepidocrocite (γ-FeOOH), goethite (α-FeOOH), magnetite (Fe3O4) and amorphous phases such as ferric oxy-hydroxides or ferroxyhyte ((-FeOOH). The study of morphology and structure of the oxide layer complements the understanding of the properties of the corrosion products, since the more compact and dense the higher the anticorrosion protection layer is obtained[5,6]. In the initial stages of the formation of the oxide layer lepidocrocite (γ-FeOOH) is formed which is subsequently transformed into goethite (α-FeOOH), which is the thermodynamically stable phase. After that the corrosion rate of the steel is considerably reduced[5]. The formation of these phases has been explained by different corrosion mechanisms, where intermediate species of short duration are formed[7].
Several researchers have investigated the relationship between the hydrodynamics of the medium and the steel corrosion rate. Studies have shown that when the steel is subjected to a hydrodynamic system the corrosion kinetics of the steel is increased[4]. It has been verified that for the carbon steel-chloride system in a slightly acidic medium, the Levich model expresses adequately the relation between the cathodic current and the mass transport during the metal corrosion[3]. However, when sulfate is added to the solution, the Levich relationship is not satisfied due to the formation of a porous layer of corrosion products. Several models have been proposed to simulate the experimental behavior of the mass transport process, taking into account both the mass transport and the different reactions that may occur during the corrosion of steel in acid sulfates[8]. The fact is that the formation of corrosion products during the anodic dissolution of the steel and the influence of the mass transport in the anodic and cathodic reactions make it difficult to obtain kinetic parameters for the estimation of the actual corrosion rate by electrochemical tests.
In some studies, the kinetics of the steel dissolution processes have been quantified by plotting the Tafel lines at polarization potentials where the curve does not show a well-defined linearity due to the mass transport control occurs simultaneously[9,10]. Similarly, sometimes a Tafel region is assumed at potentials close to the OCP potential (ΔE˂90mV) which does not meet the minimum criteria to be considered in the activation control zone, because at these potentials the reverse reaction still coexists[11-13]. Situations such as those described above lead to erroneous results in the evaluation of kinetic parameters and corrosion rates, which are evidenced by comparative studies of the dissolution kinetics of steel using polarization curves, mass loss tests and chemical evaluations[14]. On the other hand, when the Tafel criteria for the calculation of the corrosion current through polarization curves are well applied, the values of corrosion rates obtained by different methods are consistent. The criteria for the correct estimation of the Tafel slopes and the respective extrapolation method were stablished more than 5 decades ago by Stern and Geary[15].
The aim of this paper is to evaluate the carbon steel corrosion rates at different aeration conditions in acid sulfate electrolyte determined by electrochemical polarization measurements, taking into account the influence of the corrosion products formation and the hydrodynamic conditions of the system. This is intended to provide a more appropriate method for estimating the actual steel corrosion rate.
2. Materials and methods
2.1. Materials
The electrochemical tests were carried out in a conventional three-electrode cell using a low carbon steel sample (AISI-SAE 1006) as working electrode with composition showed in Table 1. The working electrode was manufactured as a rotary disk (RDE) with an exposed area of 0.31 cm2 and lateral insulation with epoxy resin. Before exposure, the working electrode was polished to sand 2000, then washed with distilled water and ethanol. A large surface area platinum mesh was used as the counter electrode and a mercury-mercurous sulfate electrode (Hg / Hg2SO4) was used as reference electrode. The test electrolyte was prepared using analytical grade reagents K2SO4 (Merck) and H2SO4 (Carlo Erba) and doubly distilled water. The test electrolyte consisted of 0.2 mol L-1 K2SO4 solution. The pH of the electrolyte was set at pH = 3 by adding 0.2 mol L-1 H2SO4. In the deaerated tests, the system was sealed and nitrogen was injected into the solution during the stabilization of the open circuit potential (OCP) and during the all polarization test. Before initiating the polarization measurements, the system was left in rest until reaching the OCP, it took about 30 min. The cathodic and anodic polarizations were performed independently, starting in both cases from the OCP to overpotential of +/- 400mV. The sweep speed for potentiodynamic polarizations was 1.0 mV s-1.

2.2. Characterization of the surface by in-situ Raman spectroscopy
In situ Raman spectroscopy measurements were performed during the anodic polarization of the steel at different exposure times in order to determine the main corrosion products formed during the anodic dissolution of the metal. The in situ RAMAN measurements were performed in a cell (20 mL) of three electrodes using a carbon steel bar as the working electrode, leaving the circular area exposed to the electrolyte at 1 mm from the surface of the solution. A 304 stainless steel mesh was used as the counter electrode and a mercury-mercurous sulfate electrode (Hg / Hg2SO4) was used as the reference electrode. The working electrode was polarized at 200 mV of anodic overpotential with respect to OCP, which is an anodic polarization sufficient for the formation of the corrosion products. Immediately after applying the anodic overpotential, the laser was focused on the surface of the electrode. Raman spectroscopy measurements were performed using high resolution Labram HR model from Horiba Jobin Yvon under the following conditions: a focal length of 800mm, CCD detector with 1024x256 pixel resolution, laser of He / Ne 632.81 cm-1, D1 filter, hole 1000μm, A sweep range of 100-1600 cm-1 and a Nikon model BX41 microscope with X50 objective.
3. Results and discussion
3.1. Effect of aeration on the polarization curves of carbon steel in sulfate acid medium
The Butler-Volmer equation describing the current/potential relationship in an electrochemical equilibrium system or its equivalent relationship to a metal corrosion process where individual reactions of oxidation and reduction occur at steady-state without the influence of mass transport processes can be described by Eq. (1):
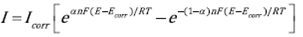 (1)
(1)
Where I is the observed current density; I corr is the current density at the corrosion potential; E is the applied potential, E corr is the corrosion potential; ( is the transfer coefficient; n is the number of electrons transferred in the reaction; R is the ideal gasses constant; T is the temperature and F is the Faraday's constant. When the anodic or cathodic reaction rate is negligible with respect to the other, a linear correlation between the potential and the logarithmic of the current is established and Eq. (1) is reduced to the well-known Tafel law, Eq. (2):
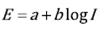 (2)
(2)
Where a and b are constants. The constant b is known as the Tafel slope, where b=(a is the Tafel slope for the anodic reaction and b=(c is the Tafel slope for the cathodic reaction. When E = E corr , then I = I corr , which is the basis for the Tafel extrapolation method to obtaining the corrosion current of the system.
The reduction of oxygen, Eq. (3), is the reaction that governs the cathodic process during the iron dissolution in slight acid and neutral sulfate solutions. However, when the electrolyte is highly acidic, e.g. pH values below 2, the hydrogen evolution reaction, Eq. (4), is the preponderant. Figure 1 shows the potentiodynamic polarization curves of carbon steel in acid sulfate electrolytes, in aerated and deaerated solutions. The dashed lines correspond to the calculation of the Tafel slopes of the cathodic and anodic reactions. In the aerated system, curve 1b, by the effect of mass transport, a limit current is observed in the cathode branch of the polarization curve. This makes it difficult to extend a line that expresses the cathodic Tafel slope.
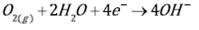 (3)
(3)
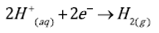 (4)
(4)
Similarly, in the anodic branch the definition of a line that expresses the anodic Tafel slope becomes difficult. This is because the rapid formation of corrosion products on the electrode surface hinders the development of a linear region in the I vs. E curve at high anodic polarization. Thus, the activation control during the dissolution of the steel is not evident. By contrast, in the polarization of the deaerated system, the linear regions of both anodic and cathodic polarizations appear well-defined. In the deaerated system the values of the calculated Tafel slopes are (c = 300 and (a = 125 mV/dec, for the cathodic and anodic reactions, respectively. Usually, values around of 100 mV/dec and 60 mV/dec have been reported for the cathodic and anodic Tafel slopes for pure iron corrosion in acid sulfate solutions[16-18]. The difference with respect to the values obtained in the present work can be due to the small quantities of alloying elements present in the steel, which can influence in a remarkable way the kinetics of the dissolution of the iron[17-19]. In the anodic region under deaerated system, the Tafel slope corresponding to the iron dissolution reaction Eq. (5) exhibits a transfer coefficient of 0.24, calculated according to Eq. (6) .
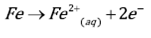 (5)
(5)
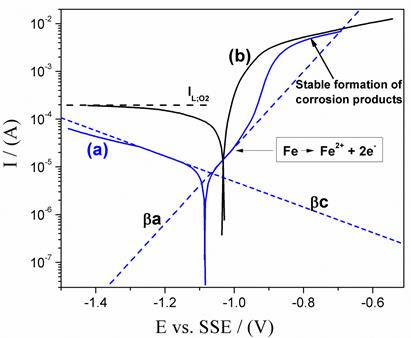
Figure 1 Potentiodynamic polarization of carbon steel in 0.2 mol L-1 K2SO4 at pH 3, T = 25 °C, rotation rate = 400 rpm, scanning speed = 1.0 mV s-1. a) deaerated, b) aerated
In anodic branch, the Tafel slope ((a) extends until approximately 100 mV of anodic overpotential. For more anodic overpotentials, the slope changes abruptly and the anodic current increases. Such anodic slope change would be related to changes in the metal dissolution mechanism, in which the formation of iron intermediary species with the oxygen or with the ions of the medium would play a main role. For very high anodic polarizations ((E > 300 mV) again, an abrupt change of the slope of the polarization curve is observed, in which the anodic current decreases due to the stable film formation of corrosion products that limit the passage of species to and from the metal surface[20,21]. The cathodic Tafel slope in the deaerated system approaches to the hydrogen evolution reaction value (Eq. (4)). The transfer coefficient for that reaction was calculated according to Eq. (7) was 0.1.
For deaerated electrolyte the anodic and cathodic transfer coefficients are calculated from the Tafel slopes, as described in Eqs (6) and (7).
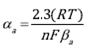 (6)
(6)
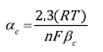 (7)
(7)
Where α c is the cathodic transfer coefficient and α a is the anodic transfer coefficient, n is the number of electrons transferred in the reaction. For the current case n = 2 for cathodic and anodic reactions. (a y (c are the anodic and cathodic Tafel slopes, respectively. R is the ideal gasses constant (8.314 J mol-1 K-1), T is the temperature in Kelvin degrees (298K), F is the Faraday's constant (96485 C mol-1).
Since the polarization curves of the steel in sulfate medium at pH = 3 under deaerated conditions clearly show linear regions at intermediate and high anodic and cathodic polarizations, following the behavior described by Tafel’s law, it is possible to determine with adequate precision the current and corrosion rate of the steel in such conditions. The corrosion current density value of the deaerated system following the extrapolation of the Tafel slopes is 18.5 μA cm-2. This value is consistent with those reported in the literature by both polarization curves (Tafel extrapolation) and weight loss methods[22].
For aerated condition, larger anodic and cathodic currents were observed. In the anodic branch of the aerated system, curve 1b, it was observed that the active dissolution process of the metal occurs with a rapid increase of the current in a narrow anodic polarization range from OCP to 100 mV of overpotential. Then, the anodic current decreases due to the formation of a stable corrosion products film on the metal surface. On the other hand, in the same aerated system, the cathode branch exhibits a diffusion limit current, whose cathodic current values are higher than those of the deaerated system over the evaluated potential range. These two processes, anodic and cathodic, of the aerated system are studied in more detail in the following sections of this work.
3.2. Anodic behavior of carbon steel in sulfate aerated medium
The anodic reaction of iron dissolution in the deaerated medium, according to that proposed by some authors, can be given by several mechanisms of reaction in which the pH and the anions of the medium play an important role in the formation of intermediary species and in the establishment of the reaction paths[18]. It has been established that at least three intermediate iron species are formed at different polarization potentials[7,18]. However, according to the study of Barcia[23], the first species formed on iron surface is always Fe(OH)ads, which is independent on other anions added in the aqueous solutions. For the other species it was proposed that sulfate and/or chloride are present in the structure of the corrosion products. The formation of different intermediate iron species at different anodic polarization conditions follows different dissolution paths, which leads to changes in the slope of the anodic branch of polarization curve[12]. However, in the aerated system the presence of oxygen completely changes the iron dissolution kinetics, stimulating corrosion and rapid formation of hydroxylated species and iron oxides. This is why the anodic currents in the presence of oxygen can be two orders of magnitude greater than free-oxyen medium at low and intermediates anodic overpotential (see curve b in Figure 1). Once the formation of stable corrosion products on the steel surface is achieved, the anodic currents in both aerated and deaerated conditions tend to be equal at high anodic polarizations. In high polarization conditions, the iron solution is controlled only by the diffusion of species through the corrosion products film formed on the metal.
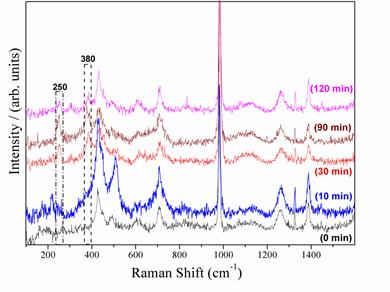
With the aim of investigating the presence of iron oxide species formed on the surface of the metal at high anodic overpotentials, measurements of in situ Raman spectroscopy were carried out on a steel electrode subjected to an anodic polarization of 200 mV with respect to the OCP. This anodic polarization condition corresponds to the overpotential in which a decreasing of the anodic dissolution rate of the metal is observed in both the aerated and deaerated systems because of the corrosion products formation. Figure 2 shows the Raman spectra of carbon steel sample under 200mV of anodic overpotential in acid sulfate solution at different times of immersion. In the Raman spectra taken at 0 and 10 min of polarization the main bands that appear in the spectra are 427.9; 509.9; 708.6; 982; 1264.5 and 1388 cm-1. These bands are associated to the sulfate electrolyte[24]. After 30 min of anodic polarization two new vibrational bands appear at 250 and 380 cm-1 which are associated to the iron hydroxide of lepidocrocite (γ-FeOOH). This stable corrosion product is formed from the reaction of iron ions in solution with dissolved oxygen and water molecules[26] [27].
3.3. Effect of hydrodynamics on the cathodic polarization of carbon steel in the aerated medium
Figure 3 shows the cathodic polarization curves of the steel in sulfate medium under aerated conditions and at different rates of electrode rotation. From the different cathodic polarization curves performed at different rotational speeds in the aerated system, a Tafel type linear region cannot be clearly identified. On the other hand, a cathodic limiting current (IL) is observed due to mass transport control, which varies according to the rotational speed of the electrode. Figure 4 shows the graph of IL vs. (1/2 which follows the behavior described by the Levich equation, Eq. (8)[8].
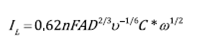 (8)
(8)
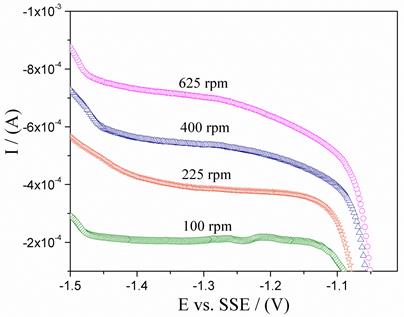
Figure 3 Cathodic polarization of carbon steel in 0.2 mol L-1 K2SO4 at different rotation speeds in aerated condition, T = 25 °C
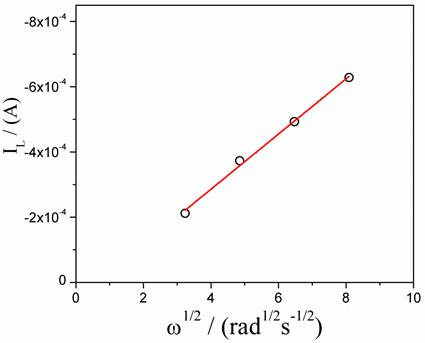
Figure 4 Cathodic limiting current as function of the rotation speed at -1.2 V(E vs. SSE) of cathodic polarization in aerated condition, T = 25 °C
Where "A" is the effective area of the electrode (0.31 cm2), "D" is the diffusion coefficient of the electroactive species (cm2 s-1), "v" is the kinematic viscosity of the fluid (8. 844x 10-3 cm2 s-1)[27], "C*" is the concentration of the electroactive species in the bulk electrolyte (mol cm-3) and "ω" is the rotation speed in rad s-1. According to Eq. (8), the slope of the straight line is directly related to the diffusion coefficient of the electroactive species. It is known that the cathodic current varies according to the aeration conditions of the system, i.e. the oxygen reduction reaction is limited by the diffusion of that species in the medium as shown in Figure 1. The oxygen concentration in the solution at a temperature of 25 °C is 2x10-7 mol cm-3[3]. The oxygen diffusivity value can be calculated as 1.24x10-5 cm2 s-1, which is consistent with the value reported in the literature[28]. This result confirms that the oxygen reduction reaction is the main cathodic reaction in the aerated sulfate medium at pH = 3 and that reaction controls the dissolution of the steel.
3.4. Corrosion rates of the steel in the sulfate aerated medium calculated from polarization curves
Taking into account that it is not possible to properly draw the Tafel slopes of the anodic and cathodic branches of the polarization curves of the steel in the sulphate medium under aerated conditions due to the absence of a linear region on the E vs. I curve at intermediates and high polarizations, the most appropriate procedure to obtain the corrosion current of the system would be to extrapolate the diffusion limit current (IL) for each hydrodynamic condition up to the line perpendicular to the OCP. This is because the oxygen reduction reaction controls the dissolution process of the steel, as was shown in the previous section, i.e. the cathodic process is controlled by concentration polarization rather than activation polarization. Moreover, at the OCP, the net rate of iron dissolution is equal to the net rate of oxygen reduction, IL.
The corrosion current density calculated by extrapolating the diffusion limit current at high rotation speed of the electrode to the OCP in the aerated system yields a value of 635 μA cm-2. Thus, the corrosion current density in the aerated system is 35 times higher than in the deaerated system. This result indicates that the oxygen present in a sulfate acid electrolyte acts as a very effective depolarizing agent and consequently drastically increases the rate of corrosion of the metal[29]. However, this does not occur in all electrolytes. McCafferty[14] studied the dissolution of carbon steel in the chlorides medium using polarization curves and colorimetric analysis, finding similar corrosion current densities by both methods with values of 25 and 30 μA cm-2 for the aerated and deaerated systems, respectively. It should be noted that McCafferty, similar to the results of the present work, obtained well-defined cathodic and anodic Tafel polarization curves in the deaerated system and polarization curves with diffusional control in the cathode branch in the aerated system.
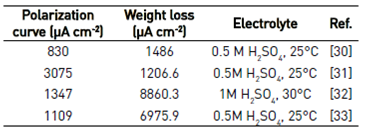
Attempting to obtain the corrosion current from the steel by extrapolating the Tafel slopes from polarization curves that do not have a clearly defined linear region (activation control) can lead to significant errors in calculating the corrosion rate of the metal. A comparison of the corrosion current densities of iron immersed in sulfate solutions calculated by polarization tests and weight loss tests is presented in Table 2. Weight loss measurements are converted to equivalent current densities using Faraday's law for comparison purpose. In general, the magnitude of the corrosion current from polarization tests is lower than that calculated by the weight loss tests, due to the fact that the Tafel criteria are not met to define correctly the linear region in the polarization curves. The results showed in the Table 2 indicate that for a correct calculation of the corrosion rate from electrochemical measurements, it is necessary to take into account other processes that occur under conditions of cathodic and anodic polarization, like mass transport process, parallel reactions and corrosion products formation.
4. Conclusions
Under natural aeration conditions, it is not possible to make a simple analysis of the corrosion of the low carbon steel immersed in sulfate solution from the extrapolation of the Tafel slopes, since such slopes are not well defined due to the formation of rust in the anodic region and the mass transport influence in the cathodic region. At cathodic polarization potentials and with natural aeration, there is an increase in polarization currents with respect to the deaerated system and the oxygen reduction reaction is controlled by the mass transport. The corrosion rate of low carbon steel immersed in sulfate solution at pH 3 is 35 times higher in natural aeration conditions than in the deaerated medium. Under deaerated conditions and at intermediate polarization potentials, there is a change in the dissolution mechanism of the steel. At high anodic overpotentials the rate of dissolution of the steel tends to be equal in both systems, aerated and deaerated because the corrosion of the metal is controlled by the diffusion of species through corrosion products film. The most appropriate procedure to obtain the corrosion current from polarization curves of the low carbon steel immersed in aerated sulfate solution would be to extrapolate the diffusion limit current (IL) for each hydrodynamic condition up to the line perpendicular to the OCP.

