











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
Permalink1. Introduction
Optically active epoxides are key intermediates in organic chemistry because they can undergo stereospecific ring-opening reactions or functional group transformations, giving rise to a wide variety of biologically or pharmaceutically important compounds [1]. Enantioselective epoxidation of non-functionalized olefins using Jacobsen type catalysts is one of the most widely used methods for the synthesis of epoxides within the asymmetric catalysis [2-4], because this type of catalysts has a high selectivity towards obtaining enantiomerically pure epoxides [5, 6]. However, separation of the catalyst from the reaction mixture and its further reuse are still problematic because the reaction product and the catalyst are in the same phase [7, 8].
To solve this problem, heterogeneous catalysts have been developed by anchoring on polymeric supports [9] inorganic solids of various characteristics [10-16] ionic liquids [17,18] nanoparticles [19] or intercalation in clays [20]. Although in all these methodologies it has been possible to retain the catalyst in the solid matrix, some examples have been found in which this immobilization process results in a significant loss of activity, asymmetric induction capacity and selectivity due to unexpected changes in the stereoelectronic behavior of the catalyst [19].
Besides this, one of the problems that is frequently questionable in the immobilization of a catalyst in inorganic supports, is the leaching of the catalyst due to the instability of the support-catalyst bond [21]. It is well-known that the reuse of an immobilized catalyst is simpler with respect to its homogenous counterpart, however, in some cases this can necessarily be preceded by the reactivation of the catalyst, when this is deactivated either by poisoning, fouling, thermal degradation or volatilization of the active components [19,22].
An alternative strategy to the immobilization and that is inside the methods of heterogeneization, is the precipitation of homogeneous catalysts by manipulating their solubility in the reaction medium [20]. One way to manipulate the solubility is to increase the molecular weight of the catalyst, facilitating product isolation and catalyst recovery, which in turn aids post epoxidation [23].
On the other hand, the oxidizing systems commonly used for this type of reactions are based on the use of iodosylbenzene, sodium hypochlorite and meta-chloroperoxybenzoic acid. These yield good results (conversions of 99% and enantiomeric excesses between 43-66%) [24, 25]; however, they promote deactivation of the catalyst because of the oxidative degradation of the salen ligand [26-28]. Additionally, when working in biphasic conditions, it is necessary to use of nitrogen coordinating co-catalyst axial bases to transfer the oxidant from the aqueous phase to the organic phase [7]. As alternatives, some oxidants such as molecular oxygen, hydrogen peroxide and dimethyldioxirane have been explored [3]. The most environmentally desirable oxidants, such as oxygen (O2) and hydrogen peroxide (H2O2) have not been effective as sources of oxygen. With O2, deactivation of the catalyst occurs by oxidative decomposition [29], whereas with H2O2 decomposition is favored in O2 and H2O [30].
Dimethyldioxiranes (DMD) have rapidly grown into one of the most useful oxidation processes in the arsenal of synthetic chemists [31]. DMD is a non-metal organic oxidant that possesses the unique ability to transfer oxygen atom to many functional groups. It has several advantages over other oxidation methods, high yield, cheap, environment friendly, it works at room temperature or low temperature [31]. As results of the enantioselective epoxidation reactions with dimethyldioxirane have been found high enantioselectivities, up to 93% and 92%, for the case of some chromenes and isoflavones, respectively [32, 33] without degrading the catalyst because it improves the stability of the catalyst thanks to the moderate oxidation conditions that can be attained [34].
Bearing in mind the problems of recovery of the catalyst and the oxidative power of the commonly used oxidants, in this article the results obtained in the enantioselective epoxidation of styrene are reported using DMD as an oxidizing agent and dimeric homochiral Mn(III)-Schiff base catalysts without immobilization.
2. Experimental procedure
2.1 General
Styrene, 3-tert-butil salicylaldehyde, trioxane, 3,5-di-tert-butil salicylaldehyde, (1R,2R)(- diaminocyclohexane, N-Methylmorpholine-N-Oxide (NMO) and Pyridine-N-Oxide (PyNO) were supplied by Sigma-Aldrich. NaHCO3, toluene chloroform and dichloromethane were purchased of Merk. Na2SO4 and methanol were acquired of J. T. Baker. Finally, glacial acetic acid and ethanol were acquired of Fluka and Panreac, respectively. Thermo Scientific Nicolet is50 FT-IR Spectrometer with attenuated total reflectance was used for obtaining the infrared spectra. Thermo Scientific Evolution 300 UV spectrometer was used for acquired the UV-vis spectra. Varian CP-3800 chromatographic equipment was used for quantification of the reaction products using a B-Dex chiral capillary column (length 30 m, I.D 0.25 mm, film thickness 0.25 µm).
2.2 Catalyst synthesis
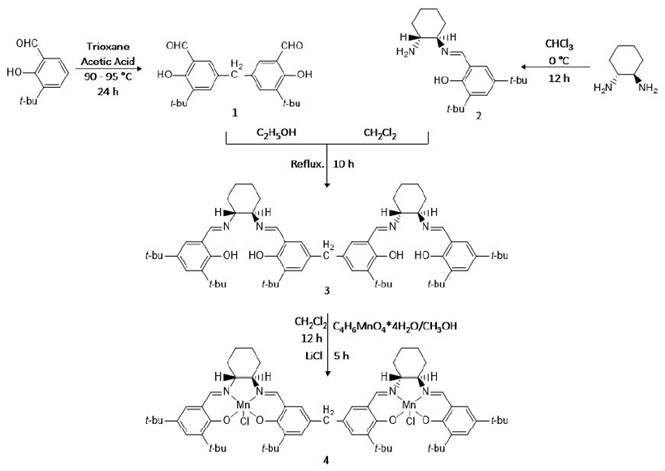
The synthesis procedure of dimeric homochiral Mn(III)-Schiff base was the result of the combination of the following steps: first, synthesis of the dialdehyde ligand [36], then synthesis of the Schiff base ligand [37] and finally the synthesis of the dimeric ligand and the catalyst according the method established by Kureshy et al [35].
Synthesis of 5,5-methylene-di-3-tert-butyl salicylaldehyde (Dialdehyde Ligand) 1
Synthesis of dialdehyde was carried out with a minor modification to the reported method by Janssen et al [36]. In a typical experiment, a solution of 3-tert-butyl salicylaldehyde 73.8 mg (4.14 mmol) and trioxane 11.7 mg (1.3 mmol) in 10 ml of glacial acetic acid was heated to a temperature of 90-95 °C under nitrogen atmosphere. A mixture of concentrated sulfuric acid and glacial acetic acid (0.32/0.68 ml/ml) was dropwise added. The temperature of resulting solution was maintained for 24 h. Subsequently, the reaction mixture was poured into cold water and allowed to stand overnight. The precipitated solid was filtered and extracted with hexane (30 ml). The solvent was removed under vacuum at 68 °C, resulting in dialdehyde as a reddish yellow solid in 40% yield.
Synthesis of N-(2-hydroxy-3,4-di-tert-butyl benzaldehyde)-1-amino-2-cyclohexeneimine (Schiff Base) 2
Synthesis of the chiral Schiff base was carried out with a minor modification to the reported method [37]. In a typical procedure, 3,5-di-tert-butyl-2-hydroxybenzaldehyde (3.02 mmol) and (1R,2R)(−)diaminocyclohexane (3.02 mmol) were stirred in chloroform at 0 °C for 12 h, and then the mixture was warmed up to room temperature. The solvent was removed under vacuum at 60 ºC and unreacted starting materials were removed by washing with water leading to the formation of the “half unit” Schiff base as a yellow solid in 84% yield.
Synthesis of 5,5-methylene di-[(R,R)-{N-(3-tert-butyl salicylidine)-N’-(3’,5’-di-tert-butyl salicylidene)}-1,2-cyclohexanediamine] (Dimeric Ligand Schiff Base) 3
A solution of 5,5-methylene-di-3-tert-butyl salicylaldehyde (0.478 mmol) in ethanol was mixed with Schiff Base (0.956 mmol) in dichloromethane. The resulting mixture was refluxed for 8-10 h. The excess solvent was removed by vacuum and then precipitated out the chiral ligand as a greenish-yellow solid in 80% yield.
Synthesis of 5,5-methylene di-[(R,R)-{N-(3-tert-butyl salicylidine)-N’-(3’,5’-di-tert-butyl salicylidene)}-1,2-cyclohexanediaminato (2-) manganese(III) chloride] (Dimeric Catalyst) 4
0.20 mmol of dimeric ligand Schiff base dissolved in dichlorometane was mixed with 0.40 mmol of manganese acetate tetrahydrate dissolved in methanol under an inert atmosphere (N2). The reaction mixture was stirred for 12 h, and subsequently, was cooled to room temperature. Subsequently, 0.40 mmol of lithium chloride was added and the resulting mixture was stirred while exposed to air for 5 h. The solvent was removed under vacuum and the evaporation residue was recrystallized in dichlorometane leading in homochiral dimeric catalyst as a brown solid in 90% yield. A general scheme of the synthesis of dimeric catalyst is summarized in Figure 1.
2.3 Catalyst characterization
The precursors and the synthesized catalyst were characterized by infrared spectroscopy in the region of 450 cm-1 to 4000 cm-1 and UV-Vis spectra were recorded in the range 200-700 nm.
2.4 Catalytic test
The enantioselective epoxidation of styrene using in situ generated dimethyldioxirane (DMD) as the oxidizing agent and dimeric homochiral Mn(III)-Schiff base was carried out varying olefin/oxidant molar ration, catalyst loading, reaction temperature and presence of nitrogen coordinating co-catalysts. The initial reaction conditions were 0.17 mmol KHSO5 in 8 ml of water, 0.17 mmol styrene, 1 mol% catalyst, 17 °C, absence of co-catalyst and 15 min of reaction time. For all reactions, two solutions were used. Firstly, a buffer solution (aqueous NaHCO3, 5% w/w), for keeping the pH in a range of 7.8-8.3 values. This is because the pH control during the catalytic tests is important since the formation of DMD by reaction between acetone and KHSO5 takes place in a slightly basic pH range. The second solution was the amount required of KHSO5 in form of Oxone® (2KHSO5•KHSO4•K2SO4) dissolved in 8 ml of distilled water. The reaction time was determined by the time of addition of the second solution (approximately 15 min), time in which the reaction was stopped. Subsequently, the reaction mixture was centrifuged in order to remove the catalyst containing solid. To the free solid liquid phase, 5 mL of dichloromethane were added obtaining two phases: aqueous and organic (reaction products). The aqueous phase was discarded off. The organic phase was concentrated and dried with anhydrous Na2SO4. The reaction products were analyzed by gas chromatography (GC) with flame ionization detector (FID) and using toluene (15 µl) as internal standard. The conditions of the method used were helium as mobile phase at 33 psi, split ratio = 100:1, injector and flame ionization detector temperature at 280 °C, heating program: 50 °C for 1 min, from 50 °C to 80 °C at heating rate of 2 °C/min, 80 °C for 2 min, from 80 °C to 200 °C at heating rate of 10 °C/min, 200 °C for 12 min.
3. Results and discussion
3.1 Catalyst characterization
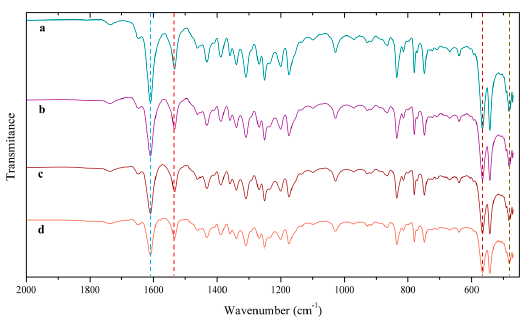
In Figure 2, the infrared spectrum of the dialdehyde ligand evidences the “bridge”, in form of methylene, that connects the two aromatic rings by the presence of the peak between 760 and 650 cm-1 and its overtone in 1740 (dotted black line) [38]. This signal remains present in the spectrum of the dimeric ligand and the dimeric catalyst. Both the Shiff base and the dimeric ligand Schiff base show a band (dotted red line) around 1630 cm-1, which is associated with the vibrations of the imine group (C=N) [8].
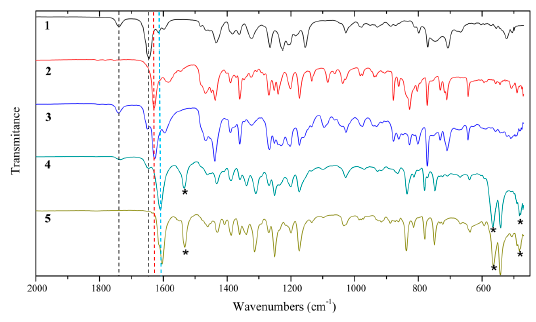
Figure 2 FT-IR spectra of (1) Dialdehyde Ligand, (2) Schiff Base, (3) Dimeric Ligand Schiff Base, (4) Dimeric Catalyst, (5) Monomeric Catalyst. *bands corresponding to the structural changes generated by the complexation of the metal
This band in the catalyst dimeric is displaced towards 1610 cm-1 (dotted blue line) evidencing the complexation of Mn by dimeric salen ligand. Furthermore, the bands located at 485, 570 and 1535 cm-1 corresponding to the bonds Mn-N, Mn-O and C-O are related with the complexation of the manganese atom [8]. On the other hand, it is observed that the dimerization of the Jacobsen catalyst does not generate additional vibrational effects besides the band corresponding to the “bridge” between the two aromatic rings, in comparison to commercial monomeric catalyst, which confirms the success of the synthesis [39].
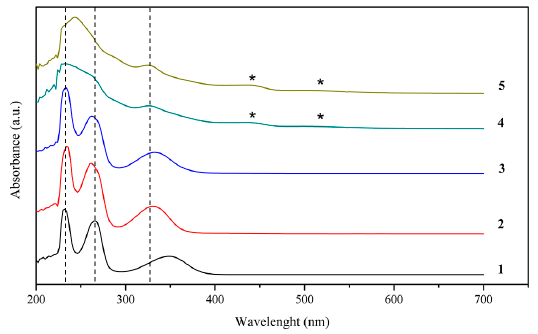
In Figure 3, the three ligands show bands that are close to 230 nm corresponding to transitions σ→σ*, close to 262 nm that are attributed to transitions π→π * and about 334 nm belonging to electronic transitions n→π * [8]. In the case of the dimeric catalyst two additional bands were observed, the first of them at 436 nm corresponding to metal-ligand charge transfers due to the complexation of the manganese atom, and the second of them at 510 nm attributed to electronic transitions d → d in the metal atomic orbitals [40]. As in the infrared spectra, in the ultraviolet spectra is observed that in comparison with the monomeric catalyst, the dimerization does not generate additional electronic transitions [39].
3.2 Catalytic activity
The dimeric homochiral Mn(III)-Schiff base complex catalyst was evaluated in the enantioselective epoxidation of styrene using in situ generated DMD. A scheme of this reaction is outlined in Figure 4.
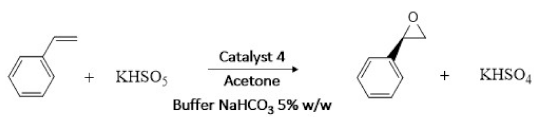
Figure 4 Enantioselective epoxidation reaction of styrene using DMD generated in situ and catalyst 4
The enantiomeric excess is determined from chromatographic areas of the 2 enantiomers (styrene oxides) by using the following Equation (1):

In this case, the majority enantiomer is the styrene R-oxide. In gas chromatography, the detector response (areas under the curve) is proportional to the amount of analyte in the volume thereof, that is, to the concentration of the analyte. Therefore, concentrations are replaced by areas and this Equation (2) is used.
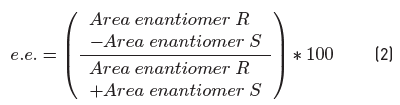
The conversion increases as the styrene/KHSO5 molar ratio increases [Figure 5]. Because the oxidant dissociates and forms dimethyldioxirane when it comes in contact with acetone, there is a non-catalytic oxidation for those molecules distanced from the sterogenic center of the catalyst. For this reason, the increment in conversion is higher than the enantiomeric excess of the product.
The increase in the enantiomeric excess of the product by increasing the catalyst loading is due to the decrease in the non-catalytic oxidation route of styrene [Figure 6]. The latter is due to a higher number of molecules of catalyst in the reaction, which allows a greater interaction between the incoming substrate and the catalytically active center. On the other hand, it is observed that the increase of the catalyst loading, from 2 to 5 mol%, does not have any considerable effect on the enantiomeric excess of the products.
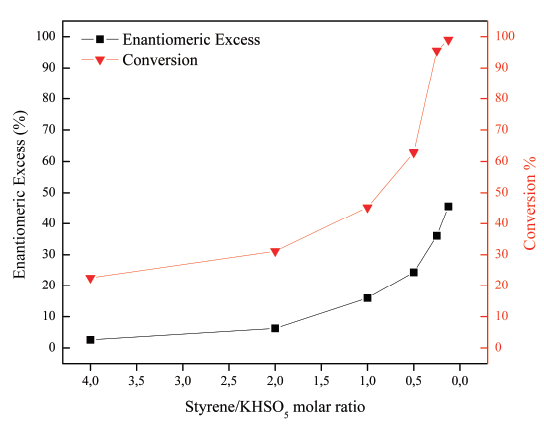
Figure 5 Effect of the styrene/KHSO5 molar ration in the enantioselective epoxidation of styrene. Reaction conditions: 8 ml acetone, 0.17 mmol styrene, 1 mol% catalyst, 8 ml water with amount of KHSO5 corresponding (dependent of styrene/KHSO5 molar ration), reaction time of 15 min and temperature of 17 °C
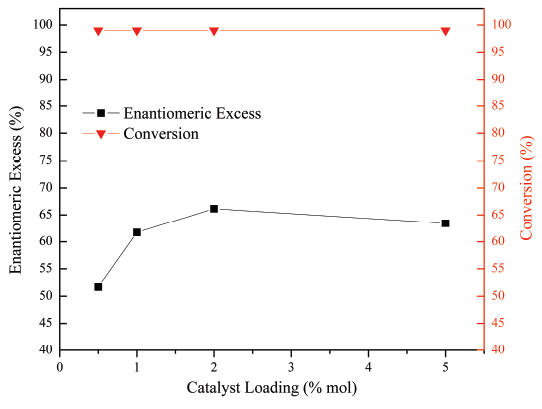
Figure 6 Effect of catalyst loading in the enantioselective epoxidation of styrene. Reaction conditions: 8 ml acetone, 0.17 mmol styrene, 8 ml water with 1.36 mmol KHSO5, 15 min and 17 °C
As it is shown in Table 1, the presence of NMO or PyNO does not affect remarkably the catalytic activity. These compounds are used as oxygen transfer agents applied to the oxidant source from the aqueous phase to the organic phase. In this case, the oxidant is generated in the reaction medium (acetone-water homogeneous mixture), and therefore it is not expected to have any beneficial effect on the reaction.
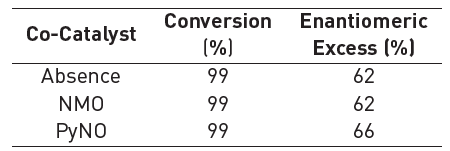
*Reaction conditions: 8 ml acetone, 0.17 mmol styrene, 2 mol% catalyst, 8 ml water with 1.36 mmol KHSO5, 15 min and 17°C.
At decreasing temperature, low conversions were obtained [Figure 7] due to decreasing kinetics energy. A constant amount of oxidant and low reaction kinetics result in an increment of non-catalytic epoxidation by the dissociation of KHSO5. It is well-known that in this reaction, the key point is the stereo-electronics interactions between the incoming substrate and catalytically active center [41]. At elevated temperatures (25°C), this type of interactions is not effective due to the increase of the reaction rate, resulting in total conversions of the substrate, but low enantiomeric excess of epoxides.
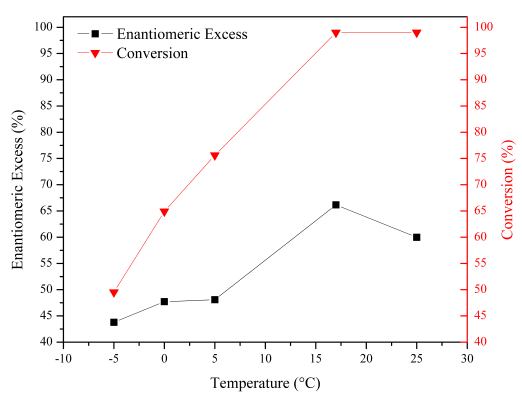
Figure 7 Effect of reaction temperature in the enantioselective epoxidation of styrene. Reaction conditions: 8 ml acetone, 0.17 mmol styrene, 2 mol% catalyst, 8 ml water with 1.36 mmol KHSO5, absence of co-catalyst and a reaction time of 15 min
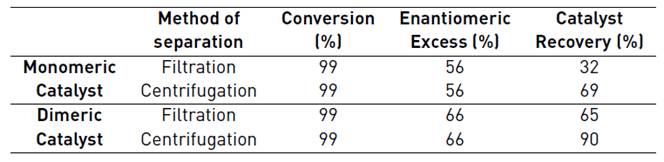
Table 2 shows that (1) regardless of the catalyst used (monomeric or dimeric) centrifugation is the best method of catalyst separation, this is because that the catalyst remains visibly evident in the filter paper used, (2) the dimeric catalyst is easier to recover than the monomeric catalyst due to the decrease of its solubility which in turn is given by molecular weight increase. Also, as expected, there is not a difference in reaction results.
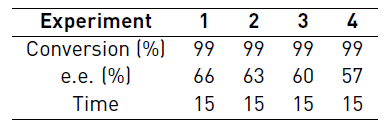
The Table 3 shows that in the catalyst reusability experiments there is a loss in the enantiomeric excess of the epoxides. The previous was attributed to physical losses of the catalyst during the separation process since the infrared spectrums of the reused catalyst do not reveal any type of oxidative degradation (see Figure 8]. However, since the amount of catalyst recovered each time is less, there is a decrease in the intensity of signals in the spectrum.
It was found that in absence of catalyst, a similar conversion than the catalyzed reaction was reached. However, the enantiomeric excess decreases significantly (3 % e.e.) because there is no sterogenic center (catalyst) "directing" the reaction towards a single enantiomer. On the other hand, no reaction was observed in absence of KHSO5.
4. Conclusions
The dimeric homochiral Mn(III) Schiff base complex 1, catalyzed successfully the enantioselective epoxidation of styrene. The best results (enantiomeric excess of 66 %) were obtained using 2 mol% of catalyst, a styrene/KHSO5 molar ratio of 0.125, a reaction temperature of 17 °C and absence of a co-catalyst. Increasing the molecular weight of the Jacobsen’s catalyst decrease its solubility, promoting the separation of reaction products and the catalyst reusability. The recovered catalyst was reused up to three times with a negligible loss of the catalytic activity due to the lost physics of the catalyst during the process of catalyst recovery. However, the catalyst did not present oxidative degradation thanks to the controlled exposition to an oxidant medium by the dosage of the diluted KHSO5. In this way, mild reaction conditions were found to increase the stability of the catalyst and to drive a truly heterogeneous catalytic process.

