










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkCES Medicina
Print version ISSN 0120-8705
CES Med. vol.25 no.2 Medellín July/Dec. 2011
REPORTE DE CASOS
Epidermólisis ampollosa en un recién nacido, reporte de un caso
Epidermolysis bullosa in a newborn, a case report
MARTHA CECILIA TORRES1, CATALINA CONTRERAS2, MARTHA LUCÍA GONZÁLEZ3
1 Fellow de Neonatología Universidad CES, Cali, Colombia. marticatorres@hotmail.com
2 Residente de II año de Pediatría, Universidad CES, Cali, Colombia
3 Dermatóloga, Fundación Valle del Lili, Cali, Colombia
RESUMEN
La epidermólisis ampollosa es una enfermedad caracterizada por la ausencia congénita parcial o total de la piel. La epidermólisis ampollosa consiste en la formación de ampollas llenas de líquido sérico o hemático. Tiene diferentes formas de presentación y el diagnóstico definitivo se hace mediante microscopía electrónica e inmunofluorescencia. El diagnóstico molecular permite descubrir el gen afectado. La epidermólisis ampollosa es una enfermedad que puede causar gran letalidad en los recién nacidos, ya que no hay tratamiento curativo definitivo; sin embargo, éste se debe enfocar en la prevención de traumas mecánicos y la curación de las ampollas, así como del manejo multidisciplinario para prevenir las complicaciones asociadas. Se presenta el caso de un recién nacido quien ingresó a la Fundación Valle del Lili de la ciudad de Cali, Colombia.
PALABRAS CLAVES
Epidermólisis ampollosa, Neonato, Colombia, Reporte de caso
ABSTRACT
Epidermolysis bullosa is a congenital disorder characterized by the partial or total loss of the skin. Epidermolysis bullosa is characterized by the formation of blisters in the skin filled of blood or fluid content. It has different clinical presentations and its diagnosis is made by electronic microscopy and inmunofluorescence technique. The molecular diagnosis makes possible to identify the affected gene. Epidermolysis bullosa is an illness that has high mortality rates in newborns, since there is no definitive cure; however, its management should be focus on the prevention of mechanical traumas, and blisters healing, therefore multidisciplinary management is necessary to prevent the complications. This is a review of a newborn's case in Fundación Valle del Lili, Cali, Colombia
KEY WORDS
Epidermolysis bullosa, Newborn, Colombia, Case report
INTRODUCCIÓN
La epidermólisis ampollosa (EA) es una entidad infrecuente en nuestro medio, caracterizada por la aparición de ampollas con contenido sérico o hemático de manera localizada o generalizada, con predominio de aparición en la mucosa de la vía aérea, boca, esófago, conjuntivas y mucosa vesical (1). Fue descrita inicialmente por Tilbury Fox en 1879 en un artículo de Lancet, que menciona el caso informado en 1875 por Hutchinson y afirma que la ausencia congénita de piel parcial o total ha sido evidenciada desde tiempos de Hipócrates (2).
Esta enfermedad aparece en todas las razas, con una incidencia de 7,7 casos por cada millón de habitantes, con un discreto predominio en el sexo masculino y con un modo de herencia que puede ser dominante o recesiva, según su clasificación (1). Se asocia a mutaciones en los genes responsables de la síntesis de las queratinas basales y de acuerdo a los genes comprometidos, las lesiones ampollares se forman a nivel de las células basales, la lámina lúcida o las capas profundas de la epidermis (1). Se han descrito alrededor de 13 genes estructurales entre la epidermis y la membrana basal y 30 clases de fenotipos diferentes, que debido a la fragilidad mecánica del tejido epitelial pueden desarrollar ampollas y heridas que no cicatrizan.
El objetivo de este reporte de caso es brindar información acerca de esta enfermedad; así como aportar bases clínicas para su clasificación, diagnóstico y manejo al personal de salud que en su práctica se ve enfrentado a pacientes con esta enfermedad.
CASO CLÍNICO
Se trata de un recién nacido de sexo masculino y cinco días de vida, quien ingresa a una institución de salud con historia de lesiones ampollosas generalizadas en tronco y cara, asociadas a marcado deterioro de su estado general (Fotos 1-2). Debido al compromiso dérmico, ingresa deshidratado, por lo cual se realizó reposición de volumen a través de catéter umbilical inicialmente y se estableció pronto inicio de nutrición parenteral con alto aporte de aminoácidos. De igual manera se instaura antibioticoterapia inicial con esquema de vancomicina más piperacilina tazobactam, que posteriormente fue cambiado a meropenem, en razón a hemocultivo positivo para Enterobacter cloacae sensible a dicho fármaco.
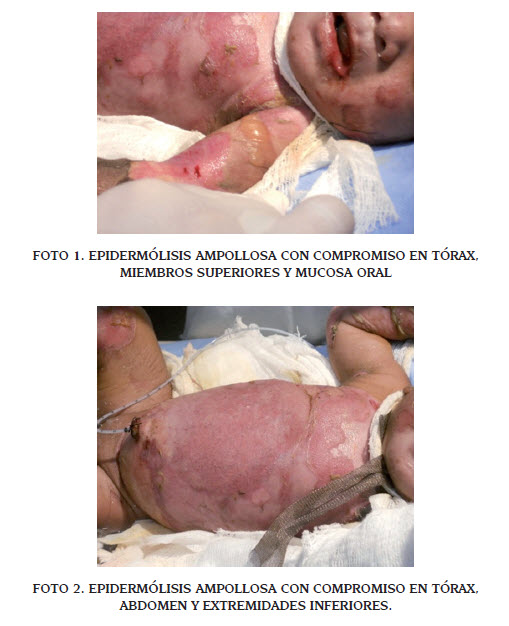
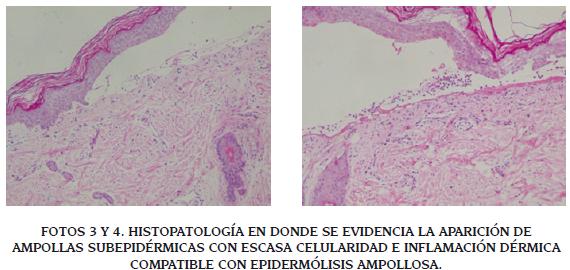
En los exámenes de ingreso se encontró pancitopenia y coagulopatía, las cuales requirieron manejo con factor estimulante de las colonias de granulocitos y transfusión de hemoderivados. Se realizó biopsia de las lesiones y se continuó manejo con curaciones y cuidados de la piel. Al tercer día de hospitalización el paciente presenta deterioro clínico, requiriendo soporte ventilatorio e inotrópico, sin embargo el paciente fallece. El reporte de biopsia de piel muestra ampollas subepidérmicas con escasa celularidad e inflamación dérmica compatible con EA (Fotos 3 y 4). No fue posible realizar microscopía electrónica.
REVISIÓN DE LA LITERATURA
El espectro de la EA incluye desde formas ampollares simples hasta una forma letal con compromiso generalizado, de acuerdo a su severidad (3). La EA puede clasificarse de acuerdo a su nivel de separación, si se encuentra en la capa basal de queratinocitos o en la capa suprabasal (4,5), de la siguiente manera:
EA simple: Es la presentación más común, y su transmisión es de tipo dominante (6). Se asocia a mutación en los genes que codifican para la queratina 5, 14 o para la plectina, y generalmente producen ampollas intraepidérmicas que curan sin cicatriz posterior. Esta incluye varios subtipos (3,6):
•Weber-Cockayne: desarrollo en la juventud, con compromiso en manos y pies.
•Dowling-Meara: caracterizado por numerosas lesiones diseminadas, principalmente con distribución herpetiforme y presente en las primeras semanas de vida (1).
•EA simple de Koebner: aparición precoz, con ampollas generalizadas y formación de quistes de millium (3).
•A nivel histopatológico, en la EA simple se observa como primer cambio, la vacuolización de los queratinocitos basales (citólisis) y por inmunoperoxidasa se puede evidenciar en el suelo de la ampolla, la tinción positiva para queratina y el colágeno IV o laminina (3).
EA de la unión: se hereda de forma autosómica recesiva, y puede representar el 1 % de los casos (3). Se encuentra compromiso de la lámina lúcida, en donde hay una formación anormal de los componentes de los hemidesmosomas, lo que da lugar a roturas a nivel de dicha estructura. Puede cursar con afectación de las mucosas (ocular, oral, faríngea, esofágica, y/o urinaria). Dependiendo de las manifestaciones clínicas, la EA de la unión puede fraccionarse en diferentes clases como lo son:
•EA Herlitz: mortal, con compromiso de las mucosas desde el nacimiento. En la microscopía electrónica hay ausencia de la proteína laminina 5 (3).
•EA no Herlitz o leve: con mutaciones de laminina 5 y fenotipo con ampollas diseminadas de menor severidad.
•EA de la unión con atresia pilórica: asociada a mutaciones en los genes que codifican la formación de la plectina (7).
EA distrófica: representa el 46,5 % de los casos. Se caracteriza por una alteración del colágeno tipo VII de las capas más profundas de la epidermis. Su herencia puede ser de forma autosómica dominante (manifestaciones leves) o recesiva (manifestaciones leves a severas) con lesiones que comprometen los dedos de las manos como de los pies y cicatrizan con deformaciones y contracturas en las áreas de flexión. Puede complicarse con infecciones secundarias (bacterianas o micóticas), anemia, alteraciones hidroelectrolíticas, compromiso nutricional y deformidades de manos y pies (1,3). A su vez, se clasifica en varios subtipos:
•EA distrófica dominante de Cockayne-Touraine (hipertrófica) y de Pacini (atrófica): producen lesiones leves en las extremidades.
•EA distrófica recesiva de tipo Hallopeau-Siemens: es una de las formas más severas, presenta entre otras manifestaciones fusión de los dedos de las manos y pies, estenosis esofágica y erosiones en la córnea. Su complicación es el desarrollo de carcinoma escamocelular, con alto riesgo de metástasis a edades tempranas (3).
•EA no Hallopeau-Siemens: tiene una forma de presentación más leve.
A nivel histopatológico, en la EA distrófica se observa una anormalidad en las fibras de anclaje, compuestas por colágeno tipo VII, que puede deberse a una anormalidad morfológica o numérica de dichas fibras.
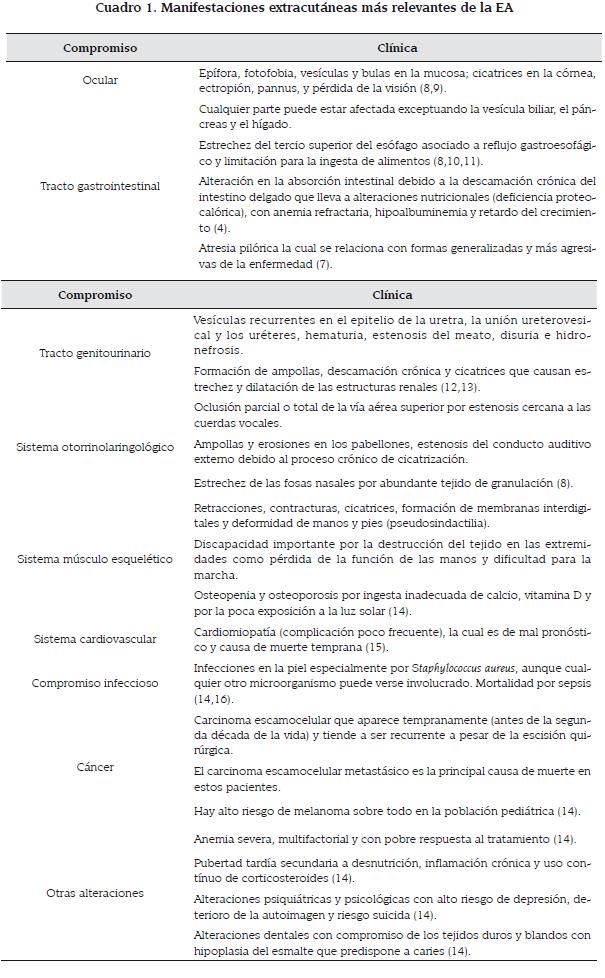
Muchos de los subtipos de la enfermedad se asocian a manifestaciones extracutáneas que aumentan la morbilidad y en ocasiones llevan a la muerte, las más comunes se destacan en el cuadro 1.
DIAGNÓSTICO
A pesar de los avances en la genética molecular de la enfermedad, el diagnóstico inicial se sigue haciendo por una combinación entre las características clínicas, la historia familiar y el nivel de localización de las lesiones.
La biopsia de piel es usada para definir el nivel de la lesión. Se realiza idealmente por afeitado de la ampolla y se debe asegurar que haya suficiente contenido dérmico. Se toma especialmente de aquellas lesiones que tengan más de 12 horas de evolución y contengan líquido serohemático (15).
La microscopía electrónica tiene como objetivo demostrar el nivel de separación de la ampolla con relación a las capas de la piel. Es difícil realizar el diagnóstico ya que pueden aparecer varios niveles de compromiso en la misma muestra o en diferentes lesiones en el mismo paciente. Si las lesiones se encuentran a nivel intraepidérmico es compatible con EA simple. Si están a nivel de la lámina lúcida es más probable que se trate de una alteración de la unión dermoepidérmica. Si la alteración está por debajo de la lámina densa está relacionado con la variedad de EA distrófica.
Cuando se encuentran hemidesmosomas pequeños es compatible con EA de la unión. Si hay fibrillas de anclaje disminuidas en número, corresponde a la variedad distrófica. El hallazgo de cuerpos estelares densos intraepidérmicos es más frecuente en la presentación neonatal. Mediante este método no es posible identificar entre las formas dominantes y recesivas (15).
Para el mapeo antigénico se usa la tinción inmunohistoquímica indirecta. Consiste en teñir la zona de unión dermo-epidérmica usando anticuerpos contra proteínas que se expresan en la piel de los pacientes con la enfermedad y en las personas sanas para detectar el nivel de la lesión. Los anticuerpos usados son: anti BP230 (tiñe la superficie inferior de las células basales que contienen los hemidesmosomas), anti queratina 14 (tiñe el citoplasma de las células epidérmicas basales), anti colágeno tipo IV (tiñe la lámina densa de la membrana basal), anti laminina - 1 (tiñe la lámina lúcida superior) (15,17).
El uso de anticuerpos específicos es un método diagnóstico de mucha utilidad, especialmente si se usa junto con la microscopía electrónica y el mapeo antigénico, pues muestra la reducción o la ausencia de expresión de ciertos antígenos en la unión dermo-epidérmica, lesiones típicas de los subgrupos de la unión (anti BP-180, 19 DEJ-1) y en la variedad distrófica (LH 7.2) (15,18).
El diagnóstico por medio del ensayo genético molecular es de fundamental importancia, pues es el método ideal para diferenciar entre los diferentes subtipos de la enfermedad y determinar la necesidad de realizar consejería genética y definir el pronóstico. También puede identificar el origen genético (si es dominante o recesivo en la variedad distrófica) y es de gran utilidad para realizar el diagnóstico prenatal (15,19).
TRATAMIENTO
Este es básicamente paliativo e idealmente preventivo. Se debe tener en consideración el manejo del dolor agudo en los pacientes ya que es una parte importante del espectro clínico, aunque hay pocos reportes en la literatura sobre el uso de estos medicamentos especialmente en los neonatos. Varios fármacos se usan actualmente incluyendo sedantes, analgésicos y anestésicos (20-22). Se debe informar a los pacientes sobre el tipo de prendas de vestir y calzado que deben usarse (23), al tiempo que se instruira sobre las actividades de riesgo que deben evitarse, ya que pueden motivar la aparición de nuevas ampollas.
Se ha descrito el uso de corticoides y retinoides con resultados variables en la forma distrófica recesiva de Hallopeau-Siemens aunque faltan más estudios. Se recomienda vigilancia médica para descartar y tratar precozmente las complicaciones oncológicas (24).
Es importante el manejo multidisciplinario por fisioterapeutas y ortopedistas para ayudar a mantener el rango completo de movimiento en las articulaciones y así minimizar las contracturas y poder reparar las deformidades de la mano. El cirujano plástico puede encargarse de realizar injertos de piel para las áreas ulceradas o desnudas. Se requiere de dilataciones de esófago, en caso de presentar estenosis y seguimiento por dermatología para realizar la extirpación de cualquier tipo de lesiones causadas por el carcinoma escamocelular (25).
Es fundamental el manejo nutricional con aporte rico en calorías y proteínas necesarias, las cuales pueden ayudar a la recuperación en éste tipo de pacientes (15,26). Se encuentra en investigación la terapia de proteínas y la terapia genética, así como el manejo con interferón sin evidencia clara aún sobre su efecto (15).
El manejo debe ser multidisciplinario y especialmente dirigido para prevenir complicaciones. Debe haber acompañamiento por parte de pediatría, dermatología, fisioterapia, ortopedia, cirugía plástica, cirugía pediátrica, nutrición, odontología, psiquiatría y psicología entre otras (27).
Hasta el 87 % de los pacientes mueren en el primer año de vida en los casos severos de EA de la unión, mientras que en la infancia, la causa más común de muerte es el carcinoma escamocelular metastásico (28).
CONCLUSIONES
Dada la gravedad de esta enfermedad y su asociación a varias comorbilidades, es pertinente realizar un diagnóstico certero temprano para iniciar el tratamiento oportuno de estos pacientes. Las diferentes formas de EA evolucionan hacia la cronicidad, con un pronóstico más reservado en los casos de tipo distrófico. No hay tratamiento curativo definitivo para estos pacientes, por lo tanto el manejo se debe enfocar a la prevención de traumas mecánicos, curación de las ampollas y cuidados especiales de la piel. Requiere manejo multidisciplinario para prevenir las complicaciones cutáneas y extracutáneas. Siempre debe incluirse el consejo genético en pacientes de riesgo e incluir asesoría psicológica al paciente y a las familias en donde se presenta esta enfermedad.
CONFLICTOS DE INTERESES
Ninguno
AGRADECIMIENTOS
Al personal médico y de enfermería de la Unidad de Cuidado Intensivo Neonatal de la Fundación Valle del Lili por la colaboración que nos brindaron para la elaboración de este trabajo, así como al Dr. Edwin Carrascal quien nos colaboró con la toma de microfotografías de las biopsias del paciente.
REFERENCIAS
1. Muñoz H. Epidermolisis bulosa. En: Hubner ME, Ramírez R, Nazer J, eds. Malformaciones congénitas, diagnóstico y manejo neonatal; 2005.Pp. 471-474. [ Links ]
2. Rosset M. Case Reports: Epidermolysis bullosa. Canad M.A.J. 1956;75:507-509. [ Links ]
3. Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner Tuderman L, Heagerty A, et al. The classification of inherited epidermolysis bullosa (EB): Report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol 2008;58(6):931-950. [ Links ]
4. Uitto J, Richard G. Progress in epidermolysis bullosa: from eponyms to molecular genetic classification. Clin Dermatol 2005;23:33-40. [ Links ]
5. Miranda A, Frías G. Hierro S. Epidermólisis ampollosa. Revisión Clínica. Rev. Mex Pediatr 2003;70(1):32-36. [ Links ]
6. Horn HM, Tidman MJ. The clinical spectrum of Epidermolysis bullosa simplex. Br J Dermatol 2000;142(3):468-472. [ Links ]
7. Natsuga K, Nishie W, Shinkuma S, Arita K, Nakamura H, Ohyama M, et al. Plectin deficiency leads to both muscular dystrophy and pyloric atresia in Epidermolysis Bullosa Simplex. Mutat Hum 2010;31(10):1687-1698. [ Links ]
8. Fine JD, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa. Part I. Epithelial associated tissues. J Am Acad Dermatol 2009;61:367-384. [ Links ]
9. Fine JD, Johnson LB, Weiner M, Stein A, Cash S, DeLeoz J, et al. Eye involvement in inherited epidermolysis bullosa (EB): experience of the National EB Registry. Am J Ophthalmol 2004;138:254-262. [ Links ]
10. Fine JD, Johnson LB, Weiner M, Suchindran C. Gastrointestinal complications of inherited epidermolysis bullosa: cumulative experience of the National EB Registry. J Pediatr Gastroenterol Nutr 2008;46:147-158. [ Links ]
11. Freeman EB, Koglmeier J, Martinez AE, Mellerio JE, Haynes L, Sebire NJ, et al. Gastrointestinal complications of epidermolysis bullosa in children. Br J Dermatol 2008;158:1308-1314. [ Links ]
12. Kajbafzadeh AM, Eimi A, Mazaheri P, Talab SS, Jan D. Genitourinary involvement in epidermolysis bullosa: clinical presentations and therapeutic challenges. BJU Int 2010;106(11):1763-1766. [ Links ]
13. Fine JD, Johnson LB, Weiner M, Stein A, Cash S, DeLeoz J, et al. Genitourinary complications of inherited epidermolysis bullosa: experience of the national epidermylosis bullosa registry and review of the literature. J Urol 2004;172:2040-2044. [ Links ]
14. Fine JD, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa Part II. Other organs. J Am Acad Dermatol 2009;61:387-402. [ Links ]
15. Fine JD, Burge SM. Genetic Blistering Diseases. En: Burns T, Breathnach S, Cox N, Griffiths C, eds. Rook's Textbook of Dermatology; 2010 Pp. 1857-1894. [ Links ]
16. Tyring SK, Chopra V, Johnson L, Fine JD. Natural killer cell activity is reduced in patients with severe forms of inherited epidermolysis bullosa. Arch Dermatol 1989;125:797-800. [ Links ]
17. Frew JW, Dopping-Hepenstal P, McGrath JA. Categorizing immunofluorescence mapping in epidermolysis bullosa with pyloric atresia: Use as a broad prognostic indicator. Australas J Dermatol. 2010;51(3):212-214. [ Links ]
18. Chen M, Chan LS, Cai X, O'Toole EA, Sample JC, Woodley DT. Development of an ELISA for rapid detection of anti-type VII collagen autoantibodies in epidermolysis bullosa acquisita. J Invest Dermatol. 1997;108(1):68-72. [ Links ]
19. Has C. Molecular genetic assays for inherited epidermolysis bullosa. Clin Dermatol 2011;29(4):420-426. [ Links ]
20. Loepke AW, Soriano SG. An assessment of the effects of general anesthetics on developing brain structure and neurocognitive function. Anesth Analg 2008;106:1681-1707. [ Links ]
21. Anand K, Aranda JV, Berde CB, Buckman S, Capparelli EV, Carlo WA. Analgesia and anesthesia for neonates: study design and ethical issues. Clin Ther 2005;27(6):814-843. [ Links ]
22. Herod J, Denyer J, Goldman A, Howard R. Epidermolysis bullosa in children: pathophysiology, anaesthesia and pain management. Paediatr Anaesth 2002;12(5):388-397. [ Links ]
23. Gannon BA. Epidermolysis bullosa: pathophysiology and nursing care. Neonatal Netw. 2004;23(6):25-32. [ Links ]
24. Herrera E, Sanz A. Bosch RJ. Epidermólisis ampollosa hereditaria. [Sitio en Internet] Hallado en URL: http://www.e-dermatosis. com/pdf-zip/Derma039.pdf Acceso en enero 11 de 2011. [ Links ]
25. Chuan-Hong K, Sue-Jen C, Betau H, An Hang Y, Chih Yi H, Cheng Hung H. Junctional Epidermolysis Bullosa. J Chin Med Assoc 2006;69(10):503-506. [ Links ]
26. Baquero C, Herrera E, López JC, De Lucas R, Romero J, Serrano MC, et al. Guía de atención clínica integral de la epidermólisis bullosa hereditaria. Ministerio de Sanidad y consumo 2008. [Sitio en Internet] Hallado en URL: http://www.msps.es/profesionales/prestacionesSanitarias/publicaciones/docs/epidermolisisBullosa.pdf Acceso en octubre 19 de 2009. [ Links ]
27. Fine JD, Johnson LB, Weiner M, Suchindran C. Cause-specific risks of childhood death in inherited epidermolysis bullosa. J pediatr 2008;152(2):276-280. [ Links ]
28. McGrath J, Mellerio JE. Epidermolysis bullosa. J Med Archives 2011; 24(1):74-88. [ Links ]
Recibido en: septiembre 20 de 2011. Revisado: agosto 15 de 2011. Aceptado: octubre 9 de 2011.
Forma de citar: Torres MC, Contreras C, González ML. Epidermólisis ampollosa en un recién nacido, reporte de un caso. Rev CES Med 2011; 25(2):221-230

