










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkCES Medicina
versión impresa ISSN 0120-8705
CES Med. vol.26 no.2 Medellín jul./dic. 2012
Facomatosis pigmentovascularisasociada a síndrome deKlippel-Trenaunay y a alopeciatriangular. Reporte de caso
Pigmentovascularis phacomatosis associated to Klippel-Trenaunay syndromeand triangular alopecia. A case report
MANUELA CADAVID, FRANCHEZCA ZAPATA1, NATALIA VELÁSQUEZ2
1 Residentes de dermatología CES, franchezca@gmail.com
2 Dermatóloga pediatra, docente Universidad CES
RESUMEN
La facomatosis pigmentovascularis se caracteriza por la coexistencia de malformacionesvasculares cutáneas con nevus melanocíticos y hasta en un 50 % de los casos puedetener compromiso sistémico. El sistema de clasificación tradicional identificaba cinco categorías,numeradas del I al V de acuerdo a la presencia de cierto tipo de nevus, subdivididos en tipo a y bdependiendo de si existe o no compromiso sistémico. Más recientemente, Happle propone una nuevacategorización basada en los hallazgos clínicos así: cesioflammea (mancha mongólica y nevusflammeus), spilorosea (nevus de Spilus y nevus telangiectásico) y cesiomarmorata (mancha mongólicay cutis marmorata telangiectásico congénita), siendo la primera la más común. Se presentael caso de un paciente masculino de tres años de edad, quien desde el nacimiento presentaba una malformación vascular capilar tipo nevus flammeus extenso,mancha mongólica, melanosis escleral e iris mamilado,una malformación venosa congénita localizadaen la región suprapúbica, además de alopecia triangular,a quien se le hizo el diagnóstico inicial de facomatosispigmentovascularis tipo cesioflammea y posteriormentese asoció a síndrome de Klippel Trenaunay.
PALABRAS CLAVE
Facomatosis pigmentovascularis, Síndrome de Klippel Trenaunay Weber, Malformaciones vasculares, Estudios de caso
ABSTRACT
Phacomatosis pigmentovascularis is a disordercharacterized by cutaneous vascular malformationsassociated with melanocytic nevi: up to50 % of patients have systemic involvement. Thetraditional classification system identifies 5 categoriesnumbered from I to V according to thepresence of certain types of nevi, and it subdividesthe categories into type a and b, dependingon whether or not systemic involvementis present. More recently, Happle proposed anew classification based on clinical findings asfollows: cesioflammea (Mongolian spot and nevusflammeus), spilorosea (telangiectatic nevusand nevus Spilus) and cesiomarmorata (Mongolianspot and congenital telangiectatic cutismarmorata), the first one being the most commonone. We present a case of a 3 year old malepatient who presented at birth with a capillaryvascular malformation, an extensive nevus flammeus,a Mongolian spot, scleral melanosis andmammilated iris, a suprapubic congenital venousmalformation and triangular alopecia; hewas diagnosed with cesioflammea phacomatosispigmentovascularis that was later associatedwith Klippel Trenaunay Syndrome.
KEY WORDS
Phacomatosis pigmentovascularis, Klippel Trenaunay Weber Syndrome, Vascular malformations, Case reports
INTRODUCCIÓN
En 1982, John Mulliken y Julie Glowacki propusieronla categorización biológica de las anomalíasvasculares, basándose en las característicaspatológicas del endotelio predominante y suprogresión natural (1).
Esta clasificación fue aceptada en 1996 por laISSVA (por sus siglas del inglés: Internacional Societyfor the Study of Vascular Anomalies), la cual catalogalas anomalías vasculares en dos grandesgrupos: los tumores (como los hemangiomas),que a menudo están ausentes o son pequeñosal nacimiento, crecen rápidamente en la infanciatemprana durante los primeros seis mesesde vida, con un máximo crecimiento al año deedad, involucionan en la niñez y nunca aparecenen la adolescencia o edad adulta, su etiologíadesconocida implica un mecanismo de proliferacióncelular endotelial.
El segundo grupo corresponde a las malformacionesvasculares, cuyo origen radica en una alteracióno error del desarrollo y formación de loscanales vasculares, siendo, por tanto, procesosbenignos que están presentes al nacer, crecencon el niño, nunca involucionan y a menudo seexpanden (1-4). A su vez, este segundo grupose clasifica en función de las características del flujo vascular (bajo o alto) y en función del tipohistológico del vaso (capilar, linfático, venoso,arterio-venoso y combinados) (1-4).
El nevus flammeus, también llamado mancha envino de Oporto, es una malformación vascularcapilar, que se observa como una mácula de coloraciónrosada rojiza que con el tiempo va oscureciéndose.Por lo general es congénita, peropuede no hacerse aparente hasta varios díasdespués del nacimiento. Ocurre en un 0,4 % delos recién nacidos, sin distinción entre géneros.En un 83 % de los casos se encuentra en cabezay cuello (5).
Se han reportado varios síndromes asociadosa nevus flammeus, como el síndrome de SturgeWeber, que presenta alteraciones neurológicasasociadas a nevus flammeus en la distribución delnervio trigémino; el síndrome de Cob o angiomatosiscutáneo meningoespinal, el síndromede Klippel Trenaunay y la facomatosis pigmentovascularis,siendo los dos últimos parte de lasmanifestaciones clínicas del paciente que a continuaciónpresentamos (5-7).
CASO CLÍNICO
Se trata de un niño de tres años, quien fue llevadoa la consulta de urgencias por presentarcuadro de vómito, dolor y distensión abdominalde 10 días de evolución. Traía una ecografía abdominalque le habían realizado en su entidadde salud, que mostraba hepatomegalia, sin lesionesfocales. El servicio de pediatría realiza undiagnóstico inicial de hemangiomas cutáneosasociado a posible compromiso visceral que pudieraexplicar el cuadro abdominal.
Dentro de los antecedentes personales, la madrerefería alopecia areata que era manejada conesteroides tópicos, así como lesiones en piel, detipo vascular, desde el nacimiento. Fue productode un parto vértice espontáneo a las 39 semanasy presentaba un neurodesarrollo normal.Otros antecedentes personales y familiares noeran relevantes para el caso. Se solicitó, entonces,evaluación por el servicio de dermatologíapara que emitiera su concepto sobre las lesionesen piel.
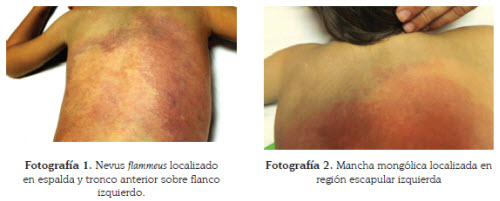
Al examen físico presentaba máculas violáceasde gran extensión, de bordes irregulares y algunastelangiectasias localizadas en espalda ytronco anterior sobre flanco izquierdo, que continuabanhacia la parte proximal del miembroinferior izquierdo (fotografía 1).
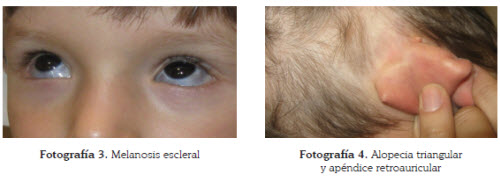
Igualmente se apreciaba una mácula grisáceade bordes mal definidos, sugestiva de manchamongólica, en región proximal de miembros superioresy región escapular izquierda (fotografía 2). En las escleras se observaban unas máculasazuladas violáceas (fotografía 3). Además, en laregión temporal izquierda y parietal del cuerocabelludo, se insinuaban algunas placas alopécicascompatibles con alopecia triangular congénita(fotografía 4).
También llamaba la atención un vaso sanguíneo,al parecer venoso, a nivel de la sínfisis del pubis,ingurgitado y doloroso a la palpación.
Con base en la historia clínica, evolución y hallazgosal examen físico, se consideró que laslesiones en piel no correspondían a hemangiomas,sino a lesiones vasculares de tipo nevusflammeus. Los diagnósticos diferenciales fueronlos siguientes síndromes que se asocian a nevusflammeus: síndrome de Sturge Weber, síndromede Cob, síndrome de Klippel Trenaunay y facomatosispigmentovascularis.
Se solicitaron exámenes paraclínicos (coprológico,hemoleucograma, función renal y hepáticay pruebas de coagulación) los cuales fueron reportadoscomo normales. La angiorresonanciade abdomen reportó un puente safeno dilatadoa nivel suprapúbico, y sin fenómeno trombóticoasociado. En la resonancia magnética nuclearde la columna dorso-lumbar y cerebral no seobservaron alteraciones. Por último, se solicitaevaluación por el servicio de oftalmología quiendiagnostica melanosis escleral e iris mamilado.
Con los hallazgos de nevus flammeus extenso,mancha mongólica, melanosis escleral e iris mamilado,alopecia triangular y malformación venosacongénita se hace el diagnóstico de facomatosispigmentovascularis tipo cesioflammea.
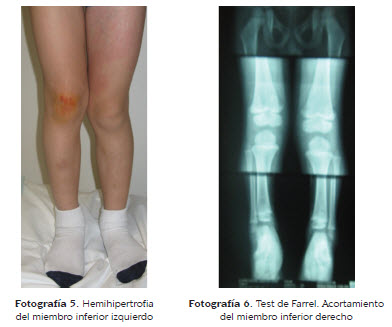
Tres meses después de la evaluación inicial, elpaciente presenta dolor en el miembro inferiorizquierdo, por lo cual le realizan ecografíadoppler a color, encontrando trombosis venosacrónica oclusiva del segmento iliaco izquierdo,la cual es manejada con anticoagulante subcutáneo.Se le realiza, además, angioresonancia dela extremidad afectada, encontrando asimetríasecundaria a hemihipertrofia del miembro inferiorizquierdo (fotografía 5).
Al examen físico se encuentra perímetro delmuslo derecho de 29 cm y del muslo izquierdode 31 cm. Se le realiza prueba de Farrel, en laque se evidencia un acortamiento del miembroinferior derecho de 9 mm de longitud (fotografía6). Con esto último se confirma el diagnósticode síndrome de Klippel Trenaunay asociado a lafacomatosis pigmentovascularis ya mencionada.
DISCUSIÓN
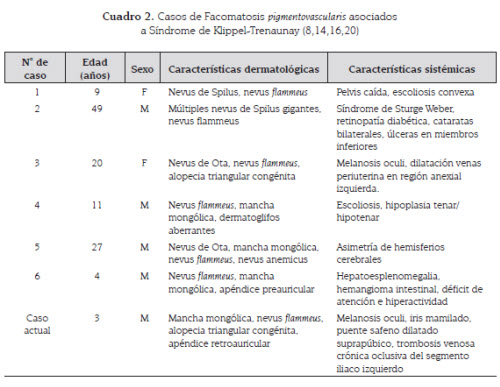
La facomatosis pigmentovascularis se define comola coexistencia de malformaciones vascularescutáneas con nevus melanocíticos, siendo losmás comunes la melanocitosis dérmica o manchamongólica, el nevus de Spilus y la melanocitosisocular. Fue descrita por primera vez en1947 por Ota y desde entonces han sido descritospocos casos. Este autor propuso una clasificaciónen dos tipos: tipo I, nevus flammeus ynevus pigmentosus et verrucosus y tipo II, nevus flammeuscon manchas mongólicas aberrantes (8).
En 1966, describió la coexistencia de nevusflammeus y nevus spilus como tipo III. El tipo IVfue descrito por Hasegawa y Yasuhara, en 1979,como una combinación de nevus flammeus, manchasmongólicas aberrantes, nevus spilus y nevusanemicus; a su vez, estos cuatro subtipos sesubdividían en a y b, dependiendo de su asociacióncon compromiso sistémico o no, respectivamente(8,9). Un quinto grupo ha sido descritomás recientemente e incluye cutis marmorata telangiectásicacongénita y la melanosis dérmica(10) (cuadro 1).
En el 2005 se reajustó esta clasificación y se definierontres grupos de acuerdo a la descripciónclínica: cesioflammea en la que se asocia un nevusde Celsius (mancha mongólica) a un nevusflammeus; spilorosea conformada por un nevus deSpilus y un nevus telangiectásico, y por último,la variedad cesiomarmorata, en la que coexiste unnevus Celsius y el cutis marmorata telangiectásicocongénito.

La verdadera frecuencia de la facomatosis pigmentovascularises desconocida, debido a que lamayoría de estudios provienen de reportes decaso. Se sabe que la más frecuente es la cesioflammea,la cual representa el 77 % de los casosreportados (8).
Se estima que el 50 % de los pacientes tienecompromiso sistémico asociado, el cual incluyedefectos en el sistema nervioso central, alteracionesoculares como melanosis oculi o glaucoma,agenesia renal, hamartomas y mamilacionesdel iris, alopecia, siendo los principales reportessobre alopecia triangular, anormalidades venosaso linfáticas como el síndrome de KlippelTrenaunay y el de Sturge Weber, entre otras (11-15). El síndrome de Klippel-Trenaunay se ha reportadoen un 6-8 % de los casos de facomatosispigmentovascularis (8,16) y se caracteriza porla presencia de una extensa malformación vascularvenular de tipo nevus flammeus -que afectaprincipalmente uno de los miembros inferioreshasta en un 95 % de los casos-, una malformaciónlinfática -presente en aproximadamente un50 % de los pacientes- y una malformación venosa-manifestada por venas laterales anómalas ovenas embrionarias persistentes prominentes-.La hipertrofia esquelética y aumento de tejidosblandos de uno o más miembros es otra de lascaracterísticas comúnmente encontradas eneste síndrome, el cual va haciéndose llamativocon la edad. La principal complicación del síndromede Klippel-Trenaunay es la tromboflebitis,que ocurre en el 20-45 % de los pacientes yocasiona embolismos pulmonares en el 4-25 %de los casos (5,14,16-18).
El diagnóstico de las malformaciones vasculareses clínico y raramente se requiere biopsia. El estudiode estos pacientes debe incluir eco dúplexpara clasificar la malformación vascular según lavelocidad del flujo, rayos X de estructuras óseasadyacentes a las lesiones en piel (para descartarhipertrofia y asimetrías), tomografía contrastaday resonancia magnética (para obtener informaciónanatómica y hemodinámica más precisa),y, finalmente, pruebas de laboratorio, ya queun perfil procoagulante en estos pacientes,puede predecir episodios de trombosis a repetición(19).
Hasta un 50 % de los pacientes presentan compromisosistémico, lo que exige un manejomultidisciplinario, requiriendo una evaluacióncuidadosa, seguimiento continuo y diagnósticooportuno de posibles complicaciones (19).
El paciente descrito presentaba una facomatosispigmentovascularis asociada a un síndrome deKlippel Trenaunay, lo cual de por sí es una asociaciónpoco frecuente; además, si se tiene encuenta el diagnóstico de alopecia triangular sólose encontró un reporte en la literatura que relacionelos tres hallazgos (ver cuadro 2).
Sin embargo, y a pesar de la escasa frecuenciade dichas asociaciones, el alto índice de sospecha,el manejo interdisciplinario y el seguimientoestrecho por medio de ayudas diagnósticas,nos permitieron descartar otras asociacionessistémicas y hacer un tratamiento oportuno delas complicaciones que presentó el paciente.
CONCLUSIÓN
Se presenta el caso de un niño de tres añoscon una malformación vascular capilar tipo nevusflammeus extenso, una mancha mongólica,melanosis escleral e iris mamilado, además deuna alopecia triangular y malformación venosacongénita, en quien se hizo el diagnóstico inicial de una facomatosis pigmentovascularis tipo cesioflammea,las cuales posteriormente se asociarona un síndrome de Klippel- Trenaunay. Seresalta la importancia de un examen físico detalladoy de exámenes diagnósticos de tipo imagenológicosen aquellos pacientes que presentenmalformaciones vasculares significativas al nacimientoo que se asocien a otros de los hallazgospreviamente mencionados.
BIBLIOGRAFÍA
1. Mulliken JB, Glowacki J. Hemangiomas andvascular malformations in infants and children:a classification based on endothelialcharacteristics. Plast Reconstr Surg1982;69:412-22. [ Links ]
2. Enjolras O, Mulliken JB. Vascular tumors andvascular malformations (New issues). AdvDermatol 1997;13:375-423. [ Links ]
3. Aristizábal Dávila AM, Ruiz AC, Zuluaga A.Anomalías vasculares en la infancia. RevAsoc Colomb Dermatol 2004;12:11-26. [ Links ]
4. MacFie CC, Jeffery SLA. Diagnosis of vascularskin lesions in children: an audit and review.Pediatric Dermatol 2008;25:7-12. [ Links ]
5. Redondo P. Vascular malformations (I).Concept, classification, pathogenesisand clinical features. Actas Dermosifiliogr2007;98:141-58. [ Links ]
6. Redondo P, Aguado L, Martínez-Cuesta A.Diagnosis and management of extensivevascular malformations of the lower limb. JAm Acad Dermatol 2011;65:893-906. [ Links ]
7. Ortega T, Cajone M, Pasquali P, Trujillo B,Roizental M. Malformaciones vasculares depredominio cutáneo: diagnóstico y tratamiento.Dermatol Venez 2005;43:4-11. [ Links ]
8. Fernández-Guarino M, Boixeda P, de las HerasE, Aboin S, García-Millán C, Olasolo PJ.Phakomatosis pigmentovascularis: Clinical findingsin 15 patients and review of the literature.J Am Acad Dermatol 2008;58:88-93. [ Links ]
9. Goyal T, Varshney A. Phacomatosis cesioflammea:first case report from India. IndianJ Dermatol Venereol Leprol 2010;76:307. [ Links ]
10. Torrelo A, Zambrano A, Happle R. Cutismarmorata telangiectatica congenita andextensive mongolian spots: type 5 phacomatosispigmentovascularis. Br J Dermatol2003;148:342-5. [ Links ]
11. Happle R. Phacomatosis pigmentovascularisrevisited and reclassified. Arch Dermatol2005;141:385-8. [ Links ]
12. Shields CL, Kligman BE, Suriano M, ViloriaV, Iturralde JC, Shields MV, et al. Phacomatosispigmentovascularis of cesioflammea type in 7patients: combination of ocular pigmentation(melanocytosis or melanosis) and nevusflammeus with risk for melanoma. Arch.Ophthalmol. 2011;129:746-50. [ Links ]
13. Mandt N, Blume-Peytavi U, Pfrommer C,Krengel S, Goerdt S. Phakomatosis pigmentovascularistype IIa. J Am Acad Dermatol1999;40:318-21. [ Links ]
14. Turk BG, Turkmen M, Tuna A, Karaarslan IK,Ozdemir F. Phakomatosis pigmentovascularistype IIb associated with Klippel-Trénaunaysyndrome and congenital triangularalopecia. J Am Acad Dermatol. 2011;65(2):e46-49. [ Links ]
15. Kim HJ, Park KB, Yang JM, Park SH, Lee ES.Congenital triangular alopecia in phakomatosispigmentovascularis: report of 3 cases.Acta Derm Venereol 2000 may;80(3):215-6. [ Links ]
16. Vidaurri-de la Cruz H, Tamayo-Sánchez L,Durán-McKinster C, Orozco-Covarrubias Mde la L, Ruiz-Maldonado R. Phakomatosispigmentovascularis II A and II B: clinical findingsin 24 patients. J Dermatol 2003;30:381-8. [ Links ]
17. Zea MI, Hanif M, Habib M, Ansari A. Klippel-Trenaunay Syndrome: a case report withbrief review of literature. J Dermatol CaseRep 2009;3:56-9. [ Links ]
18. Meier S. Klippel-Trenaunay syndrome: a casestudy. Adv Neonatal Care 2009;9:120-4. [ Links ]
19. Redondo P. Vascular malformations (II).Diagnosis, pathology and treatment. ActasDermosifiliogr 2007;98:219-35. [ Links ]
20. Diociaiuti A, Guidi B, Sanchez JAA, FelicianiC, Capizzi R, Amerio P. Phakomatosispigmentovascularis type IIIb: A case associatedwith Sturge-Weber and Klippel-Trenaunaysyndromes. J Am Acad Dermatol2005;53:535-8. [ Links ]
Recibido: febrero 10 de 2012. Revisado: octubre 19 de 2012. Aceptado: octubre 25 de 2012.
Forma de citar: Cadavid M, Zapata F, Velásquez N. Facomatosis pigmentovascularis asociada a síndrome
de Klippel-Trenaunay y a alopecia triangular. Reporte de caso. Rev CES Med 2012; 26(2): 229-236
