










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkCES Medicina
Print version ISSN 0120-8705
CES Med. vol.27 no.1 Medellín Jan./June 2013
Síndrome de Hajdu Cheney, una enfermedad poco frecuente
Hajdu Cheney syndrome, an infrequent pathology
LINA MARÍA COLMENARES ROLDÁN1, NATALIA DE LA CALLE2
1 Residente de segundo año de Dermatología, Universidad CES. Correo electrónico: linaco80@hotmail.com
2 Dermatóloga, Universidad CES, Medellín, Colombia.
Recibido: octubre 22 de 2012. Revisado: febrero 22 de 2013. Aceptado: marzo 5 de 2013.
RESUMEN
El síndrome de Hajdu Cheney es un desorden autosómico dominante raro, que se acompaña de acroosteólisis, múltiples manifestaciones clínicas y radiológicas, que predominan en la adolescencia y edad adulta temprana. Hasta el momento solo han sido reportados unos 60 casos en la literatura, siendo este síndrome de presentación poco frecuente. Recientemente se ha asociado con defectos genéticos específicos del gen NOTCH-2 relacionado con la osteoclastogénesis.
Siendo esta entidad poco frecuente se destaca la importancia de presentar el primer caso reportado en Colombia, en un paciente de 20 años con acroosteólisis, talla baja, úlcera plantar, pérdida prematura de dientes, y facies llamativas, y a quien se le hace el diagnóstico de síndrome de Hajdu Cheney y actualmente recibe tratamiento por un grupo multidisciplinario para el manejo de su enfermedad.
PALABRAS CLAVE
Síndrome de Hajdu Cheney, Acroosteólisis, Úlcera plantar, Resorción ósea.
ABSTRACT
Hajdu Cheney syndrome is a rare autosomal dominant disorder, which is accompanied by acroosteolysis and multiple clinical and radiological manifestations, predominantly in adolescence and early adulthood. Until now only 60 cases has been reported in the literature, this syndrome has an infrecuent presentation; recently the syndrome has been associated with specific genetic defects in Notch 2 gene related to osteoclastogenesis.
Due to this is rare disease, we highlights the importance of presenting the first case reported in Colombia of a 20 years old patient with acroosteolysis, short stature, plantar ulcer, premature loss of teeth, and characteristic facies which has been diagnosed with Hajdu Cheney syndrome and is currently receiving treatment from a multidisciplinary team.
KEY WORDS
Hajdu-Cheney syndrome, Acro-Osteolysis, Plantar ulcer, Bone, Resorption.
INTRODUCCIÓN
El síndrome de Hajdu Cheney es una enfermedad poco frecuente que se caracteriza por presentar resorción ósea distal progresiva, alteraciones craneofaciales y dentales, talla baja, entre otras; a pesar de ser una enfermedad congénita las características que se asocian al síndrome se van haciendo más evidentes con la edad y de esta forma orientan el diagnóstico (1,2).
La forma de presentación esporádica es la más frecuente y de manera reciente se ha encontrado asociación del síndrome a mutaciones en el gen NOTCH2 relacionado a procesos de osteoclastogénesis (3,4). El diagnóstico es clínico y el manejo debe ser multidisciplinario y enfocado a las diferentes alteraciones que se manifiesten, ya que no existe un tratamiento curativo específico de la enfermedad (5).
Son escasos los reportes que se encuentran de esta entidad en la literatura. Dentro de algunos de sus diagnósticos diferenciales se encuentran artritis psoriática mutilante, epidermólisis ampollosa, trauma, lepra, tromboangeítis obliterante hiperparatiroidismo, esclerodermia, sarcoidosis y porfiria (6).
A continuación se presenta el que consideramos es el primer caso reportado en Colombia de un paciente con síndrome de Hajdu Cheney y se hace una breve revisión de la literatura.
PRESENTACIÓN
Se trata de un paciente de sexo masculino, de 20 años de edad y residente en Medellín -Colombia-, quien consultó por cuadro clínico de un año de evolución que consistía en resorción del hallux derecho y de ocho meses de evolución de una úlcera dolorosa en la región plantar derecha de 4 x 5 cm de diámetro.
Como antecedentes personales presentaba hipoacusia, cicatrices en cara, tórax y extremidades posteriores a traumatismos, talla baja, escoliosis toracolumbar, pérdidas dentales prematuras. No reportó antecedentes familiares de importancia.
Durante el examen físico se evidenciaron las siguientes alteraciones: en pie derecho acortamiento de 1er, 2do y 4to dedos, con acropaquia e hiperpigmentación en dorso de pies y dedos; en planta del pie derecho presentaba úlcera de bordes irregulares pero bien definidos, de fondo limpio; en el segundo dedo del pie izquierdo se encontró una lesión incipiente en la región distal similar a la anteriormente descrita (figura 1). En extremidades superiores presentaba manos cortas, sin engrosamiento cubital, además marcada escoliosis toracolumbar y múltiples cicatrices hipopigmentadas e hiperpigmentadas. No había alteraciones de la sensibilidad en cara y cuerpo.
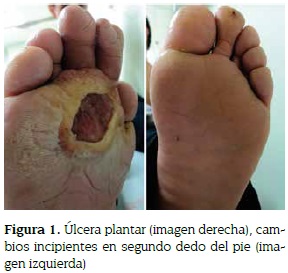
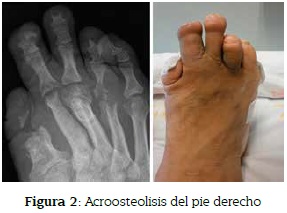
Con un diagnóstico inicial de mal perforante debido a la ulceración plantar y acroosteólisis en estudio se solicitan exámenes complementarios, encontrando en las radiografías comparativas de pies compromiso de tipo osteolítico a nivel de falange proximal y metatarsianos de 1ero, 2do, 4to y 5to dedos, de falange distal del 2do, 3ro y 4to dedo, osteopenia y solución de continuidad en los tejidos blandos en la región plantar derecha (figura 2).
En la resonancia magnética de columna se encontró escoliosis toracolumbar sin signos de compresión medular; la electromiografía evidenció polineuropatía sensitiva axonal que comprometía los nervios radiales, cubitales, medianos y surales bilaterales con retardo en las conducciones motoras; en la nasofibrolaringoscopia se encontró desviación septal sin perforaciones. Las baciloscopias realizadas fueron negativas. En la biopsia de piel se describe una ulcera inespecífica, con coloraciones de Zielh-Nielsen y Zielh-Nielsen modificado negativas, y finalmente, una reacción en cadena de polimerasa negativa para lepra.
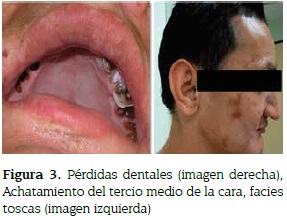
Teniendo en cuenta los hallazgos encontrados en este paciente con acroosteólisis, talla baja, pérdida prematura de dientes, facies llamativas (figura 3) y habiendo descartado otras entidades se orienta el diagnóstico a un síndrome de Hajdu Cheney.
DISCUSIÓN
El síndrome de Hajdu Cheney se caracteriza por presentar diferentes manifestaciones clínicas y radiológicas, siendo la destrucción focal ósea progresiva y la osteoporosis generalizada, las más marcadas. Fue descrito inicialmente por los doctores Hajdu y Kauntze en 1948 y posteriormente por el doctor Cheney en 1965 (1,2).
Este síndrome ha sido reportado en la literatura con diferentes nombres como síndrome de acroosteólisis, artro-dento-osteodisplasia, displasia cráneo-esquelética con acroosteólisis, osteodisplasia familiar y síndrome de Hajdu-Cheney. Es raro encontrar el fenotipo completo en la infancia, ya que por lo general este se va desarrollando a través de los años (2).
De forma reciente se ha asociado el síndrome a mutaciones en el gen NOTCH-2 el cual codifica proteínas transmembrana importantes en la señalización intercelular requeridas para el desarrollo del sistema conectivo y de otros órganos, llevando a la activación de la osteoclastogénesis y de esta forma ocasionando las diferentes manifestaciones (1,3,4).
Hasta el momento la mayoría de casos reportados han sido esporádicos, sin embargo se reconocen casos familiares con patrón autosómico dominante. A pesar de ser un desorden congénito las manifestaciones de este síndrome predominan en la adolescencia y la edad adulta temprana, y en la literatura solo se han encontrado 60 casos reportados (5).
Las osteólisis se clasifica en primarias y secundarias; las primarias se dividen en siete grupos según su patogénesis: I, enfermedad asociada a un componente neurosensorial; II, asociada a angiomatosis; III, no asociada a angiomatosis; I V, asociada a un componente cutáneo y de herencia autosómico dominante; V, acro-osteólisis multicéntrica; VI, asociada a acrogeria y progeria; VII, atrofia ósea familiar.
El síndrome de Hajdu-Cheney hace parte de las osteólisis primarias tipo II asociadas en su patogenia a angiomatosis, y esto se puede ver en la histología en donde el hueso es remplazado por pequeños vasos sanguíneos que presentan engrosamiento en la pared. Se diferencia de la osteólisis masiva o enfermedad de Gorham-Stout ya que esta última no presenta displasias óseas, presentes en el síndrome de Hajdu Cheney (6).
El diagnóstico de esta enfermedad es clínico y se hace por medio de los criterios de Brenan y Pauli (2,7):
-
Acroosteólisis
-
Huesos vormianos o el mantenimiento de las suturas craneales
-
Platibasia
-
Pérdida prematura de los dientes
-
Rostro tosco
-
Pelo grueso
-
Achatamiento del tercio medio de la cara
-
Estatura baja (<percentil 5 °)
-
Historia familiar positiva.
Se considera diagnóstico si un adulto presenta una de las tres siguientes combinaciones: acroosteólisis e historia familiar positiva de la enfermedad, o acroosteólisis y tres manifestaciones del 2 a 8 o historia familiar positiva y dos manifestaciones del 2 a 8. En niños se hace el diagnóstico presentando cuatro características del 1 a 8 o historia familiar positiva y dos características del 1a 8 (2,7). La micrognatia es el único signo identificable al nacimiento, presente en más de la mitad de los pacientes con el síndrome.
Por edades se pueden identificar diferentes características clínicas y físicas que se mencionan a continuación (2,4,8-10):
Nacimiento: micrognatia, telecantus, hipertelorismo, implantación baja de las orejas, base nasal amplia, anormalidades renales, riñones poliquísticos.
Infancia temprana: estatura baja, pelo tosco, hirsutismo, sinofris (falta de separación entre las cejas), aplanamiento del tercio medio de la cara, rostro tosco, cejas gruesas, cara ancha, hipermovilidad articular, alteraciones del lenguaje, piel seca y áspera, pérdida auditiva, alteraciones del desarrollo, infecciones recurrentes, múltiples fracturas, retraso en el desarrollo.
Infancia tardía y adolescencia: cambios acroosteo-líticos, displasia del peroné (en serpentina), disminución de la densidad ósea, acropaquias, dislocación de la cabeza del radio, alteraciones dentales.
Adultez temprana: prominencia occipital, reabsorción del hueso alveolar, dolor causado por la acroosteólisis, deformidades óseas en espalda, escoliosis, cifosis, fracturas por compresión, cefaleas.
Adultez tardía: mandíbula prominente, cambios quísticos óseos, anormalidades articulares, síntomas neurológicos.
El caso descrito presentaba acroosteólisis, achatamiento medio de la cara, estatura baja, rostro tosco, pérdida prematura de los dientes, pelo grueso, cumpliendo de esta forma con los criterios de Brenan y Pauli. Además se le encontró osteopenia, neuropatías, mal perforante plantar, cifoescoliosis, orejas prominentes e hipoacusia, también reportados en la literatura en este síndrome (4,7,9,11).
Algunos de los diagnósticos diferenciales que hay que tener en cuenta cuando se presentan manifestaciones óseas de tipo acroosteólisis son: fenómeno de Raynaud severo, exposición al cloruro de polivinilo, daño térmico, accidente ofídico, hiperparatiroidismo, esclerodermia, sarcoidosis, artritis psoriática mutilante, epidermólisis ampollosa, trauma, tromboangeítis obliterante, porfiria, síndrome de KID (queratosis, ictiosis, sordera), enfermedad de Ehlers-Danlos tipo I V, acro-osteopatía úlcero-mutilante de The-nevard, acroosteólisis asociada al síndrome de Down (7,12-15)
Algunos trastornos que causan deterioro de los nervios periféricos que puede dar lugar a acroosteólisis y se pueden confundir con el síndrome de Hajdu Cheney son la lepra, siringomelia y la insensibilidad congénita al dolor (2,7). El síndrome de meningocele lateral también podría confundirse con estadios iniciales de la enfermedad, ya que comparte algunas características con el síndrome de Hajdu Cheney como son los huesos vormianos, micrognatia, achatamiento facial, y la estatura baja, pero esta enfermedad cursa sin acroosteólisis, perdidas dentales y platibasia (16).
El manejo de esta entidad debe ser multidisciplinario y enfocado en las diferentes alteraciones que se vayan manifestando, ya que no existe un tratamiento curativo específico de la enfermedad. Un objetivo fundamental del tratamiento es evitar la osteoporosis y prevención de fracturas, por lo cual se recomienda el uso de bifosfonatos. (5)
AGRADECIMIENTOS
Los autores agradecen al Instituto Colombiano de Medicina Tropical por su colaboración.
REFERENCIAS
1. Majewski J, Schwartzentruber JA, Caqueret A, Patry L, Marcadier J, Fryns J-P, et al. Mutations in NOTCH2 in families with Hajdu-Cheney syndrome. Hum Mutat 2011 oct; 32(10):1114-7. [ Links ]
2. Brennan AM, Pauli RM. Hajdu-Cheney syndrome: evolution of phenotype and clinical problems. Am J Med Genet 2001; 100: 292-310. [ Links ]
3. Simpson MA, Irving MD, Asilmaz E, Gray MJ, Dafou D, Elmslie F V, et al. Mutations in NOTCH2 cause Hajdu-Cheney syndrome, a disorder of severe and progressive bone loss. Nat Genet 2011 abr; 43(4):303-5. [ Links ]
4. Gray MJ, Kim CA, Bertola DR, Arantes PR, Stewart H, Simpson MA, et al. Serpentine fibula polycystic kidney syndrome is part of the phenotypic spectrum of Hajdu-Cheney syndrome. Eur J Hum Genet 2012 jan; 20(1):122-4. [ Links ]
5. Hwang S, Shin DY, Moon SH, Lee EJ, Lim S-K, Kim OH, et al. Effect of zoledronic acid on acro-osteolysis and osteoporosis in a patient with Hajdu-Cheney syndrome. Yonsei Med J 2011 may; 52(3):543-6. [ Links ]
6. Iglesias-Gamarra A, Vásquez Lamadrid J, Abud Mendoza C, Quiroz F. Osteólisis. Toro G. (eds). En enfermedades metabólicas del hueso. Instituto Nacional de Salud. 1992; 21: 491-512. [ Links ]
7. Cunha I, Saavedra MJ, Oliveira MA, Salvador MJ, Malcata A. Hajdu-Cheney Syndrome: a case of acroosteolysis. Acta Reumatol Port 2007 Apr-Jun; 32(2):169-74. [ Links ]
8. Takatani R, Someya T, Kazukawa I, Nishimura G, Minagawa M, Kohno Y. Hajdu-Cheney syndrome: infantile onset of hydrocephalus and serpentine fibulae. Pediatr Int 2009 dic; 51(6):831-3. [ Links ]
9. Currarino G. Hajdu-Cheney syndrome associated with serpentine fibulae and polycystic kidney disease. Pediatr Radiol 2009 ene; 39(1):47-52. [ Links ]
10. Marik I, Kuklik M, Zemkowa D, Kozlowski K. Hajdu-Cheney syndrome: report of a family and a short literature review. Australas Radiol 2006 dic; 50(6):534-8. [ Links ]
11. August DA, Ramos DC. Anesthesia for a child with Hajdu-Cheney syndrome. Paediatr Anaesth. 2009 jun; 19(6):649-50. [ Links ]
12. Sahin A, Pepeler MS, Shimbori N. A patient with acro-osteolysis syndrome: Hajdu-Cheney. Intern Med. 2010; 49(1):87-8. Epub 2010 Jan 1. [ Links ]
13. Letchumanan P, Thumboo J, Leong RT. A patient with progressive shortening of the fingers. J Rheumatol 2009 Jan; 36(1):198-9. [ Links ]
14. Galván F, Rojas A, Méndez P, Restrepo J F, Rondón F, Iglesias A. Acro-osteólisis primaria. Un síndrome con múltiples expresiones clínicas. Presentación de un caso y análisis de la literatura. Rev Esp Enferm Metab Oseas 2002; 11:105-8. [ Links ]
15. O'Reilly MA, Shaw DG. Hajdu-Cheney syndrome. Ann Rheum Dis 1994 Apr; 53(4): 276-9. [ Links ]
16. Gripp KW. Lateral meningocele syndrome and Hajdu-Cheney syndrome: different disorders with overlapping phenotypes. Am J Med Genet A 2011 jul; 155A(7):1773-1774. [ Links ]
