










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkCES Medicina
Print version ISSN 0120-8705
CES Med. vol.27 no.2 Medellín July/Dec. 2013
Artículos de investigación científica o tecnológica
Histiocitosis de células de Langerhans en niños. Descripción de 10 casos
Langerhans cell histiocytosis in children. A description of 10 cases
ANA MARÍA GÓMEZ1, VIVIANA LOTERO2, PIEDAD MARTÍNEZ3, DIEGO MEDINA4, OSCAR RAMÍREZ3
1Residente de Pediatría. Universidad CES. Fundación Valle del Lili. Medellín-Colombia.
2Onco-Hematología pediátrica y Docente de pediatría Fundación Valle del Lili. Grupo de investigacion clínica de la Fundación Valle del Lili. Medellín-Colombia.
3Onco-Hematología pediátrica y Docente de pediatría Fundación Valle del Lili. Grupo de investigacion clínica de la Fundación Valle del Lili. Medellín-Colombia.
4Onco-Hematología pediátrica y Docente de pediatría Fundación Valle del Lili. Grupo de investigacion clínica de la Fundación Valle del Lili. Medellín-Colombia.
5Onco-Hematología pediátrica Centro Médico Imbanaco. Medellín-Colombia.
RESUMEN
Introducción: la histiocitosis de células de Langerhans constituye una entidad poco frecuente, con diferentes tipos de presentación clínica y patológica, que varían desde unisistémica y unifocal hasta compromiso de múltiples sistemas. Es consecuencia de la proliferación, acúmulo e infiltración en diferentes órganos, de células que hacen parte del sistema fagocítico mononuclear. El objetivo con la descripción de esta serie de casos es la de resaltar la importancia y sensibilizar a un diagnóstico y manejo oportuno de esta entidad, para una mejor calidad de vida y mayor sobrevida.
Métodos: se realizó un estudio retrospectivo en pacientes pediátricos diagnosticados en la Fundación Valle del Lili en un periodo de seis años.
Resultados: se registraron 10 casos de la enfermedad. No se observaron diferencias por sexo. La edad media de presentación fue de 33 meses. En el diagnóstico por inmuno-histoquímica se identificó histiocitosis de células de Langerhans con compromiso multisistemico -multifocal en cinco casos, uno con compromiso unisistémico - unifocal y otro unisistémica - multifocal y granuloma eosinofílico en los tres restantes. Cinco pacientes presentaron compromiso de órgano de riesgo y uno de ellos murió en el periodo de observación.
Conclusión: la histiocitosis de células de Langerhans es una enfermedad de compromiso sistémico muy poco frecuente en pediatría, con diversas formas de presentación clínica, lo que la hace una entidad de difícil diagnóstico y manejo. La inmunohistoquímica es la mejor forma de diagnóstico en esta entidad.
PALABRAS CLAVE
Histiocitosis, Células de Langerhans, Pediatría .
ABSTRACT
Introduction: Langerhans cell histiocytosis is a rare disease with different clinical and pathological presentation, is clinically divided into three groups: unifocal, multifocal unisystem, and multifocal multisystem Langerhans cell histiocytosis is a consequence of proliferation, accumulation and infiltration of cells that are part of the mononuclear phagocyte system in different organs. The goal with the description of this cases series is to highlight the importance and awareness to a timely diagnosis and management of this entity, for a better quality of life and longer survival.
Methods: Retrospective study in pediatric patients diagnosed in the Valle del Lili Foundation over a period of six years.
Results: In our series of cases there was no gender difference and the mean age at presentation was 33 months. In the diagnosis by immunohistochemistry identified Langerhans Cell Histiocytosis, the cases were classified as multisystemic -multifocal in 5 patients, unisystemic in 1 patients - unifocal unisystemic in 1 patients -multifocal eosinophilic granuloma in 3 patients. Of the 10 patients 5 had risk organ involvement and 1 died in the observation period.
Discussion: la histiocitosis de células de Langerhans es una enfermedad de compromiso sistémico muy poco frecuente en pediatría, de diversas formas de presentación clínica, lo que la hace una entidad de difícil diagnóstico y manejo. La inmunohistoquimica es la mejor forma de diagnóstico en esta entidad.
KEY WORDS
Histiocytosis, Langerhans cells, Pediatrics.
INTRODUCCIÓN
La histiocitosis representa un grupo de enfermedades determinadas por alteraciones de las células conocidas como histiocitos, que tienen funciones inmunológicas específicas, tales como procesamiento y presentación de antígenos (1,2).
La entidad se caracteriza por un acúmulo de dichas células, su proliferación e infiltración de diferentes órganos y tejidos, pudiendo comprometer más de un sistema al mismo tiempo, lo que la convierte en una entidad de difícil clasificación y manejo clínico (2-5).
Estas células son originadas en la médula ósea, presentando no solo manifestaciones cutáneas, pulmonares, hepáticas o ganglionares, sino además en la línea hematopoyética, lo que permite clasificarla de forma anatómica, patológica y clínica (2,6). Las células pueden migrar a sitios donde normalmente no se encuentran (p.e. cornea y sistema nervioso central), produciendo acúmulos, proliferación e infiltración de estos tejidos y en algunos casos comprometiendo órganos de riesgo (3,7).
Dependiendo del sitio de afectación la entidad es conocida de diferentes maneras: en su forma de lesiones solitarias como granuloma eosinofílico; la forma diseminada como enfermedad de Letterer- Siwe y su forma crónica como enfermedad de HandSchuller-Christian (7-9).
La afectación del sistema nervioso central ocurre principalmente en la región hipotálamo-hipofisiaria (10), presentándose diabetes insípida central como la primera manifestación clínica de esta entidad (11).
La presentación de histiocitosis de células de Langerhans es predominante en el sexo masculino, en una relación de 2:1 y la edad de presentación está entre los 5 y 15 años, siendo el tejido óseo el sistema más frecuentemente comprometido (cerca del 90 % de los casos) y la forma menos agresiva de la enfermedad (2,4,8,12).
En su interior, las células de Langerhans presentan los gránulos de Birbeck, que permite diferenciarlas de manera más objetiva por medio de tinciones de inmunohistoquímica, identificando las proteínas que constituyen los mencionados gránulos, como son los anti-CD 207 y anti-CD1, que se consideran patognomónicos de la enfermedad (1,3,13) y siendo este el método el diagnóstico más preciso.
El tratamiento varía según en la edad del paciente, extensión de la enfermedad y localización de lesiones, teniendo en cuenta para ello si la enfermedad es de un solo o varios sistemas y si hay o no compromiso de órganos de riesgo (7,9). Este estudio realiza un análisis retrospectivo, acerca de los diferentes tipos de presentación clínica e histopatológica de la histiocitosis de células de Langerhans, teniendo en cuenta variables establecidas en la población de estudio a describir.
METODOLOGÍA
Se realizó un estudio descriptivo y retrospectivo en pacientes pediátricos en el cual se tuvo en cuenta variables demográficas tales como duración de síntomas al momento del diagnóstico, edad, sexo, diagnóstico clínico e histopatológico, manejo, secuelas y tasa de sobrevida. En todos los pacientes se realizaron estudios de extensión (imaginológicos, bioquímicos y hematológicos) para establecer compromiso de órgano de riesgo, determinar extensión, sistemas comprometidos, manejo y sobrevida. Para esto se establecieron diferentes tipos de definiciones:
órgano de riesgo: compromiso de alguno de los siguientes sistemas: sistema nervioso central, hígado, bazo o sistema hematopoyético.
Compromiso multisistémico: lesión de dos o más sistemas diferentes
Compromiso multifocal: lesión radiológica en dos o más huesos, sin comprometer otros sistemas.
Compromiso unifocal o granuloma eosinofílico: lesión en un solo hueso, sin compromiso de otros sistemas.
Compromiso de sitio específico: lesiones en columna vertebral, algún hueso de la cara o cráneo.
La población de estudio estuvo constituida por todos los pacientes a quien se les hizo el diagnóstico de histiocitosis de células de Langerhans en la Clínica Fundación Valle de Lili, Cali Valle del Cauca durante el periodo comprendido entre 2007 a 2012.
El diagnóstico de histiocitosis de células de Langerhans se realizó por medio de historia clínica y ayudas diagnósticas, entre las que se incluyeron radiografía simple, escanografía y resonancia magnética, de acuerdo a órgano comprometido, y en todos los casos biopsia de la lesión en donde se confirma dicho diagnóstico.
Los datos fueron tomados de historias clínicas en la fundación Valle del Lili obtenidos de la base de datos de pacientes del servicio de onco-hematología pediátrica de la institución, en el periodo correspondiente.
Los datos fueron encontrados por medio del número de historia clínica y número de episodio del sistema electrónico SAP®, verificando las variables de interés, seguimiento y sobrevida hasta el momento de la descripción. Realizando, a su vez, doble chequeo en la base de datos de Excel diseñada para el estudio.
Se realizó revisión por parte del Comité de ética e Investigación Biomédica de la Fundación Valle del Lili, el cual fue aprobado para su respectiva publicación.
RESULTADOS
En total se encontraron 10 casos en el período de estudio. Se encontró una relación de género de 1,5:1 a favor de los hombres; con edades de presentación entre 11 meses y 10 años, promedio de 33 meses, la mediana entre el tiempo de inicio de los síntomas y diagnóstico fue de seis semanas (cuadro 1).
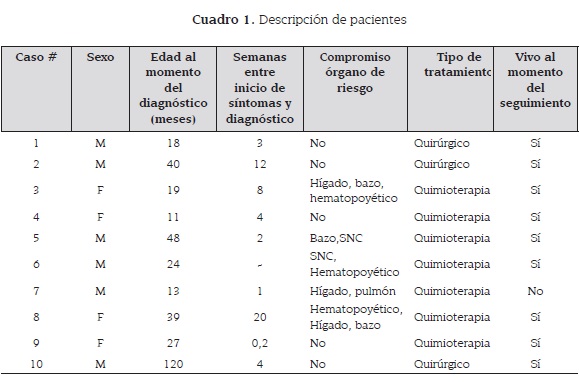
Entre las características clínicas se encontró compromiso de órgano de riesgo en el 50 % de los casos, con compromiso multisistémico y componente multifocal en cada uno de ellos; tres con compromiso en tejido óseo, dos con compromiso en sitio específico (columna vertebral y huesos de la cara).
El resto de los cinco casos que no presentaron compromiso de órgano de riesgo, tuvieron un curso favorable de la enfermedad con el manejo instaurado. Tres de los casos presentan compromiso unifocal (granuloma eosinofílico), quienes fueron manejados quirúrgicamente, en el resto de los pacientes se hizo manejo médico con quimioterapia. Ninguno de los 10 casos presentó manifestaciones dermatológicas.
Solo uno de los pacientes falleció a causa de enfermedad resistente a la quimioterapia de primera y segunda líneas. Por cuestiones administrativas el paciente había sido fue trasladado durante su tratamiento a otra institución donde falleció.
DISCUSIÓN
La histiocitosis de células de Langerhans representa un grupo raro de enfermedades de la infancia. Es de predominio en niños menores de 15 años, con un pico de incidencia entre uno a tres años de edad (7), existiendo algunos casos reportados también en población adulta. La relación hombre: mujer es de 2:1 (7).
Su incidencia varía dependiendo de la población. Es así como en las diferentes series de casos reportadas a nivel mundial puede destacarse una gran variabilidad en esta. Por ejemplo, en Estados Unidos se reporta una incidencia de 0,05 a 0,5 casos por 100 000 niños al año (7), en Francia de 0,45 casos por 100 000 niños (3) y en población latina, en donde existen pocos casos reportados, se reporta una incidencia de 0,45 casos por 100 000 al año. No se ha encontrado predilección por raza, así como componente genético (14).
La histiocitosis hace parte de un grupo de enfermedades con proliferación intensa de células que hacen parte del sistema fagocítico mononuclear (células dendríticas, monocitos, macrófagos) las cuales son originadas en la medula ósea (14,15).
Se han encontrado diferente clasificaciones, siendo la más reciente la realizada por la Sociedad Internacional de Histiocitosis: que la divide en tres tipos: Clase I como histiocitosis de células de Langerhans, Clase II histiocitosis sin células de Langerhans, en el que se incluye el síndrome hemafagocítico asociado a virus, la linfadenopatía masiva (enfermedad de Rosai-Dorfman) y el xantogranuloma infantil; y Clase III, enfermedad histiocítica maligna (sarcoma histiocítico relacionado a macrófago o célula dendrítica) (16).
Las células de Langerhans pueden presentar afectación local o sistémica, dependiendo de su proliferación y tipo de órgano infiltrado (2). Las tres variables clínicas que se presentan en la histiocitosis de células de Langerhans incluyen: una forma de afectación única con lesiones aisladas en hueso o pulmón (granuloma eosinofílico, entre el 60-80 % de los casos) de predominio en niños mayores, con pico de incidencia entre 5 a 10 años de edad; otra forma, diseminada, rara y de manifestaciones muy severas, enfermedad de Letterer- Siwe, presente en el 10 % de los casos, siendo más común en niños menores de tres años, con manifestaciones cutáneas principalmente nódulo y maculopapulares; y finalmente, su forma crónica, enfermedad de Hand-Schuller-Christian, en el 15 al 40 % de los casos, representada por la triada de lesión craneal, diabetes insípida y exoftalmos, descrita en niños entre 2 a 5 años de edad (17,18).
Sus manifestaciones clínicas dependen del sitio de afectación de la lesión, presentando un curso variable que puede manifestarse de manera unifocal o multifocal, con afectación, a su vez, de uno o más sistema (13). Una asociación importante y que define un pronóstico favorable o no, y así mismo la respuesta de quimioterapia, es el compromiso de órgano de riesgo: hígado, bazo, sistema nervioso central y sistema hematopoyético (19,20).
La manifestación unifocal en el tejido óseo representa hasta el 80 % de los casos en niños menores de cinco años, siendo el granuloma eosinofílico su principal manifestación, pudiendo comprometer casi todo el tejido óseo, excepto el de manos y pies (16).
Los síntomas van desde dolor óseo en el 80 a 90 % de los casos, inflamación de tejidos blan-dos, sensibilidad, fracturas patológicas, cefaleas (por compromiso de cráneo), disminución de la audición, procesos infecciosos como otitis o mastoiditis (compromiso de mastoides) (4), pérdidas dentales por compromiso de mandíbula y maxilar, colapso vertebral por compromiso a este nivel, hasta manifestaciones generales como fiebre, anorexia, pérdida de peso, irritabilidad, astenia y manifestaciones hemorrágicas (18,21).
Entre los estudios imaginológicos la radiografía simple permite identificar lesiones líticas medulares y reacción perióstica; la resonancia magnética nuclear es útil en delimitar la lesión y establecer la extensión local o sistémica de la enfermedad (20). La tomografía axial computarizada con emisión de positrones y la ultrasonografía son útiles para evaluar la actividad de lesiones focales, en órganos viscerales tales como hígado y pulmón, presentando la ultrasonografía ventajas en la identificación de lesiones pequeñas limitadas a tejidos blandos y adyacentes a lesiones óseas, incluyendo lesiones en cráneo (22). El diagnóstico diferencial del compromiso óseo debe hacerse entre osteomielitis y otras lesiones tumorales como el sarcoma de Ewing (18, 20).
El síndrome pulmonar, como lesión unifocal, es de presentación usual entre la tercera y cuarta década de la vida, mientras que en la población pediátrica se presenta como parte de una enfermedad multisistémica, la cual está asociada a una mayor mortalidad y en menor frecuencia a neumotórax (23).
La enteropatía perdedora de proteínas hace parte del compromiso gastrointestinal en la histiocitosis de células de Langerhans, siendo una presentación rara (menor de 1 %) y se asocia a una mortalidad de casi el 50 % (24). Cerca del 86 % de estos casos son asociados a manifestaciones cutáneas, lo que obliga a realizar diagnóstico diferencial con enteropatía perdedora de proteínas secundaria a alergia a la proteína de la leche de vaca.
Entre las manifestaciones clínicas que se presentan en la enfermedad multisistémica se encuentran síntomas constitucionales inespecíficos, con compromiso del estado general, hepato y esplenomegalia, linfadenopatías, así como citopenias con compromiso de más de dos líneas celulares (4).
A pesar de no existir una anormalidad inmunológica descrita se considera que la mayoría de estas manifestaciones son debidas a una disrupción inmunológica (25). Hasta ahora se ha logrado definir como una entidad con componente reactivo (proceso mediado por citoquinas inflamatoria) y según recientes reportes, incluirse como parte de un proceso neoplásico (por medio de marcadores inmunológicos específicos) (7,11,25).
Lo anterior ha llevado a realizar estudios anatomo-patológicos del órgano afectado y a diagnósticos diferenciales entre procesos infecciosos o malignidades hematológicas, efectuando pruebas como mielograma, biopsia de órganos afectados, inmuno-histoquímica, entre otros (6).
Para establecer el tipo de quimioterapia a realizar se debe realizar una estratificación de las diferentes categorías de riesgo, de acuerdo a la extensión de la enfermedad y grado de disfunción orgánica; igualmente se debe establecer si hay compromiso de la vida del paciente, para así definir manejos más agresivos, incluso trasplante de medula ósea (18,26).
La fortaleza del presente estudio se encuentra en determinar la población a riesgo de acuerdo a las variables mencionadas previamente. Entre las debilidades encontradas se establecen por tamaño de muestra, el cual puede ser insuficiente para identificar una adecuada incidencia de esta entidad, el paso del tiempo puede llevar a cambio de métodos diagnósticos y posibles pérdidas en el seguimiento.
CONCLUSIÓNEl objetivo con la descripción de esta serie de casos es la de recordar a los médicos generales, residentes de pediatría y pediatras que la histiocitosis, que a pesar de ser una entidad poco frecuente, puede presentarse. Su pronto diagnóstico y manejo adecuado llevan a una mejor calidad de vida y mayor sobrevida. Las diversas formas de presentación hacen que su diagnóstico sea posible si se tienen el conocimiento de que este grupo de enfermedades existe e identificando una serie de síntomas y signos clínicos que hagan sospecharla.
REFERENCIAS
1. Toro A, Restrepo R, Ochoa A. Histiocitosis de células de Langerhans. Rev Asoc Col Dermatol. 2009; 17:34-44. [ Links ]
2. Lagos-Sánchez E, Soto-Monge T, Carrillo-Henchoz JM, Suárez-Zeledón A. Síndromes histiocíticos de la infancia: a propósito de un caso de histiocitosis de células de Langerhans. Revista Médica de Costa Rica y Centroamérica. 2007; 580:167-175. [ Links ]
3. Ariza S, Cardona A, Rueda X. Histiocitosis de células de Langerhans: diez años de experiencia en el Instituto Nacional de Cancerología. Rev Asoc Col Dermatol. 2008; 16:178-184. [ Links ]
4. Arkader A, Glotzbecker M, Hosalkar H, Dormans J. Primary musculoskeletal Langerhans cell histiocitosis in children an analysis for a 3-decade period. J Pediatr Orthop. 2009: 29:201-207. [ Links ]
5. Larralde M, Abad M y Gomar B. Histiocitosis de células de Langerhans en menores de un año. Arch Argent Pediatr. 2008; 106:269-272. [ Links ]
6. Ohnishi K, Komohara Y, Sakashita N, Iyama K, Murayama T, Takeya M. Macrophages in Langerhans cell histiocytosis are diferrerentiated toward M2 phenotype: Their posible involvement in pathological processes. Pathology international. 2012; 60:27-34. [ Links ]
7. Al-Ammar A, Tewfik T, Bond M and Schloss M. Langerhans' Cell Histiocytosis: Paediatric Head and Neck Study. The Journal of Otolaryngology. 1999; 28:266-272. [ Links ]
8. Rojas R, García G, Parra D, Solar A, Oyanedel R, Díaz Fernán, et al. Compromiso óseo en histiocitosis de células de Langerhans en el niño: estudio radiológico simple: presentación clínica y diagnostico radiológico. Revista Chilena de Radiología. 2005; 11:122-128. [ Links ]
9. Márquez A, Quiceno W, Pérez JC. Histiocitosis de células de Langerhans-síndrome de Letterer-Siwe: afectación de piel, tejido linfoide, hígado, bazo y hueso. Presentación de caso. Rev Colomb Radiol. 2009; 20:2734-38. [ Links ]
10. Marchand I, Barkaoui M, Garel C, Polak M, and Donadieu J. Central diabetes insipidus as the inaugural manifestation of Langerhans cell histiocytosis: natural history and medical evaluation of 26 children and adolescents. J Clin Endocrinol Metab. 2011; 96:1352-1360. [ Links ]
11. Nagasaki K, Tsumanuma I, Yoneoka Y, Ogawa Kikuchi T, Uchiyama M. Spontaneus regression of isolated neurohypophyseal Langerhans Cell Histiocytosis with diabetes Insipidus. Endocrine Journal. 2009; 56:721-725. [ Links ]
12. Singhi A and Montgomery E. Gastrointestinal tract Langerhans cell histiocytosis: a clinicopathologic study of 12 patients. Am J Surg Pathol. 2011; 35:305-310. [ Links ]
13. Braier J.L, Goldberg J, Chantada G, Rosso D. Oncopedia. Histiocitosis. Buenos Aires: Hospital JP Garrahan. p. 2-56. [ Links ]
14. Stanton K, Schor St. G, Behrman S. Nelson textbook of pediatrics. 19th ed. New York, USA. Elsevier-Academic Press 2011; cap 501, pag 1773-1777. [ Links ]
15. Murcia J, Barcenas W. Histiocitosis. Precop 2013; 11:36-46. [ Links ]
16. Singh H, Kaur S, Yuvarajan P, Jain N, Maini L. Unifocal Granuloma of femur due to Langerhans' cell histiocytosis: A case report and review of the literature. Case Reports in Medicine. 2010; Article ID 686031.1-3. [ Links ]
17. Rodríguez Y, Navas E, Goitia O, Terán P, Ron F. Histiocitosis de células de Langerhans con afectación gástrica reporte de caso revisión de la literatura. Rev Venez Oncol. 2008. 20:91-97. [ Links ]
18. Philip Lanzkowsky. Manual of Pediatric Hematology and Oncology. 5ta ed. New York, USA. Elsevier-Academic Press 2011; cap 18, pag 567-598. [ Links ]
19. Histiocyte Society. Treatment protocol of the Third International Study for Langerhans Cell Histiocytosis, 2nd Version (LCH III protocol). Viena: Histiocyte society; 2002. [ Links ]
20. Hashmi MA, Haque N, Chatterjee A, Guha S. Langerhans cell histiocytosis of long bones: MR imaging and complete follow up studyJournal of Cancer Research and Therapeutics. 2012; 8:286-288. [ Links ]
21. Singh T, SAtheesh CT, Appaji L, Aruna BS, Mamatha HS, Giri GV et al. Langerhan's cell histiocytosis: A single institutional Experience. Indian Journal of Medical and Paediatric Oncology. 2010; 31:50-53. [ Links ]
22. Kosiak W, Piskunowicz M, Swieton D, Bien E, Batko T. Sonographic diagnosis and monitoring of localized Langerhans cell histiocytosis of the skull. Journal of Clinical Ultrasound. 2012; 41:134-39. [ Links ]
23. Khanbabaee G, Yeganeh MH, Tabatabaei SA, Khatami A, Bazrafshani S, Rezaei N. Langerhans cell histiocytosis with pulmonary involvement and unilateral pneumothorax. The Turkish Journal of Pediatrics. 2010; 52:638-641. [ Links ]
24. Shima H, Takahashi T and Shimada H. Protein-losing enteropathy caused by gastrointestinal tract-involved Langerhans cell histiocytosis. Pediatrics. 2010;125:426-432. [ Links ]
25. Rodriguez C. The Langerhans cell histiocytosis: a disease in search of an identity. Rev Bras Hematol Hemoter. 2011; 33:328-36. [ Links ]
26. Wing Tin S, Duverneuil N, Idbaih A, Garel C, Ribeiro M et al. Efficacy of vinblastine in central nervous system Langerhans cell histiocytosis: a nationwide retrospective study. Orphanet Journal of Rare Diseases. 2011; 6:2-9. [ Links ]
Recibido: julio 2 de 2013 Revisado: octubre 15 de 2013 Aceptado: octubre 23 de 2013
