










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkCES Medicina
Print version ISSN 0120-8705
CES Med. vol.28 no.1 Medellín Jan./June 2014
Artículos de revisión
Resistencia a antirretrovirales: bases moleculares e implicaciones farmacológicas
Resistance to antiretrovirals: molecular bases and pharmacological implications
DANIELA VANEGAS-OTÁLVARO1, LILIANA ACEVEDO-SÁENZ2, FRANCISCO JAVIER DÍAZ-CASTRILLÓN3, PAULA ANDREA VELILLA-HERNÁNDEZ4
1Bióloga, Estudiante de Maestría. Grupo Inmunovirología, Facultad de Medicina, Universidad de Antioquia.
2Microbióloga, DSci (c). Grupo Inmunovirología, Facultad de Medicina, Universidad de Antioquia.
3Médico, PhD. Profesor. Grupo Inmunovirología, Facultad de Medicina, Universidad de Antioquia
4Bacterióloga, MSc, DSci. Profesora. Grupo Inmunovirología, Facultad de Medicina, Universidad de Antioquia. pvelilla@gmail.com
RESUMEN
Una de las principales características del virus de la inmunodeficiencia humana es la alta diversidad genética, dada, en parte, por la baja fidelidad de la transcriptasa reversa, lo cual lleva a la generación de variantes virales con mutaciones asociadas a evasión de la respuesta inmune, cambio del tropismo celular o resistencia a medicamentos antirretrovirales. La terapia antirretroviral altamente activa es un esquema farmacológico contra el virus que utiliza dos o más familias de antirretrovirales, que pretende llevar la replicación viral a niveles indetectables, reduciendo entonces la morbimortalidad y aumentando la calidad y expectativa de vida de los individuos infectados. Las mutaciones que confieren resistencia a estos fármacos pueden ser fijadas en el genoma viral y ser transmitidas a nuevos hospederos, aportando así a la circulación de una población viral resistente a estos medicamentos que resulta en falla virológica.
En esta revisión se describen los mecanismos más comunes asociados con resistencia a antirretrovirales y las mutaciones reportadas en la literatura.
PALABRAS CLAVE
VIH, Terapia antirretroviral altamente activa, Resistencia a medicamentos, Mutación, Biología computacional.
ABSTRACT
One of the main characteristics of HIV is its high genetic diversity given in part by the low fidelity of the reverse transcriptase, which leads to the generation of viral variants bearing mutations associated with the evasion of the immune response, changes in the cellular tropism and/or antiretroviral resistance. Highly active antiretroviral therapy (HAART) is a pharmacologic scheme against HIV, which comprises two or more antiretroviral drug families, HAART aims to suppress viral replication which in turn decrease the morbidity and mortality of infected individuals, and increase their life expectancy and improve their quality of life. Mutations that confer resistance to antiretrovirals can be fixed in the viral genome and be transmitted to new hosts contributing to the movement of a viral population resistant to these drugs that results in virologic failure. In this review we describe the most common mechanisms associated with antiretroviral resistance, mutations reported in the literature and bioinformatics tools used for their determination.
KEY WORDS
HIV, Highly active antiretroviral therapy, Drug resistance,Mutation, Computational biology.
INTRODUCCIÓN
La infección por el virus de inmunodeficiencia humana (VIH) continúa siendo uno de los principales problemas de la salud pública en el mundo. A finales del 2011 se reportaron 34 millones (31,4-35,9 millones) de personas infectadas con VIH, presentándose variaciones considerables entre países y regiones (1). En el caso específico de Colombia, desde 1985 hasta el 31 de diciembre de 2011, se han notificado un total de 75 620 casos de infección por VIH, siendo la población entre 15 y 44 años la más afectada (2).
En los recientes análisis epidemiológicos se evidencia una disminución en las tasas de mortalidad asociada al síndrome de inmunodeficiencia adquirida (SIDA), al igual que en el número de infecciones nuevas. Este comportamiento en la evolución de la infección puede ser explicado, en parte, por la implementación de la terapia antirretroviral altamente activa (TARAA) (3).
Aunque esta terapia presenta múltiples beneficios en el control de la infección, también se han observado algunos efectos adversos como intolerancia ocasionada por los efectos secundarios, lo que afecta la adherencia al tratamiento; carencia de reconstitución inmunológica en algunos individuos, a pesar de tener cargas virales indetectables en plasma y, el desarrollo de resistencia a las diferentes clases de medicamentos antirretrovirales (3).
La alta tasa de mutaciones característica de este virus, genera variantes virales con resistencia a diferentes medicamentos antirretrovirales, que pueden ser seleccionadas por un proceso que involucra la presencia del medicamento dentro del microambiente celular. Se ha observado que las mutaciones asociadas a resistencia pueden ser fijadas en el genoma viral y ser transmitidas a nuevos hospederos, lo cual generará una población viral intrínsecamente resistente a uno o varios medicamentos antirretrovirales (ARV), que posteriormente se traducirá en falla virológica en este individuo (4).
OBJETIVO
El objetivo de esta revisión es describir las mutaciones y mecanismos más comunes asociados con la resistencia a medicamentos durante la terapia antirretroviral contra el VIH; además brindar una breve descripción de las herramientas bioinformáticas utilizadas para la detección temprana de la resistencia. Para el desarrollo de esta revisión, se hizo una búsqueda sistemática en las siguientes bases de datos: PubMed, Medline, Ovid y Scielo; se utilizaron los siguientes términos: "HIV-1 infection", "virologic failure", "HAART", "HIV drug resistance mutations", "antiretroviral therapy", entre otros.
TRATAMIENTO ANTIRRETROVIRAL
En los años 90, el conocimiento de los fenómenos específicos del ciclo replicativo del VIH, la interacción con el hospedero y el conocimiento de la estructura tridimensional de las proteínas virales, permitió el surgimiento de la terapia antirretroviral, como una combinación de tres o más fármacos pertenecientes a dos o más familias de fármacos antirretrovirales, la cual llevaba a una disminución en los niveles de ARN viral y a un aumento en el conteo de linfocitos T CD4+ (LT CD4+); a este tipo de tratamiento es lo que se conoce como TARAA (5). Para 2011 la cobertura de dicha terapia fue de 68 % en América Latina y 67 % en el Caribe, comparado con el 54 % de cobertura global (6).
Aunque entre 1998 y 2000 las guías de tratamiento para VIH del Departamento de Salud y Servicios Humanos de los Estados Unidos (DHHS, por sus siglas en inglés) recomendaron el uso de la TARAA para la mayoría de los pacientes, incluyendo aquellos asintomáticos y con conteos de LT CD4+ mayores a 500 cell/l, la preocupación por la aparición de virus resistentes a los medicamentos ARV y la toxicidad de los mismos llevó a que, entre 2001 y 2006, muchos programas asistenciales aplazaran el tratamiento hasta que el conteo de LT CD4+ fuera menor a 350 cell/l (7).
Se ha observado que el inicio temprano de la TARAA se asocia con un aumento en la expectativa y la calidad de vida, disminución en la probabilidad de desarrollar co-infecciones, reducción en la tasa de hospitalización (8) y de la aparición del fenómeno denominado discordancia inmunovirológica, que consiste en la poca o nula recuperación en los conteos de LT CD4+ a pesar de presentarse una supresión viral completa (9).
En contraste, diversos ensayos clínicos han demostrado que el inicio de la terapia antirretroviral altamente activa con conteos de LT CD4+ inferiores a 200 cell/µl se asocia con mayores tasas de falla virológica y resistencia a los antirretrovirales (10, 11).
Recientemente la carga viral ha sido propuesta como un factor determinante en la elección del esquema de tratamiento antirretroviral, si se tiene en cuenta que pacientes que se encuentran bajo terapia con rilpivirina y exhiben cargas virales > 100 000 copias/ml, tienen un mayor riesgo de falla virológica (12).
RESISTENCIA A LOS MEDICAMENTOS ANTIRRETROVIRALES
La diversidad y disponibilidad en fármacos antirretrovirales han permitido que en muchos individuos infectados y tratados con TARAA, se logre una supresión viral exitosa; sin embargo, en un número importante de pacientes no se logra dicha supresión, dada la aparición de cepas resistentes al tratamiento antirretroviral, lo cual puede depender de diferentes factores tanto del hospedero como del virus (17).
La no adherencia al tratamiento es la principal causa de falla virológica y ha sido fuertemente asociada con la aparición de mutaciones de resistencia específicas para las diferentes clases de medicamentos ARV (13). Una segunda infección con otra cepa de VIH, fenómeno conocido como superinfección, es otro de los factores que contribuyen a la generación de virus recombinantes que pueden llevar a la aparición de variantes resistentes a medicamentos ARV (14,15).
En el caso de los factores virales se han observado patrones de mutaciones asociados a resistencia propios de algunos tipos y subtipos virales; este fenómeno, que podríamos denominar resistencia natural, se debe a que casi todos los fármacos antirretrovirales se desarrollaron para cepas de VIH-1 subtipo B, a pesar de que sólo el 10 % de las infecciones corresponden a éste y que su distribución geográfica se limita a países de América y de Europa Occidental (16). Un ejemplo de lo anterior es la nevirapina, medicamento al cual se presentan tasas de resistencias previas al tratamiento de 69 %, 36 %, 19 % y 21 % para los subtipos C, D, A, y CRF02_AG, respectivamente (17).
La pérdida de susceptibilidad de los virus ante un agente antirretroviral es causada por una o múltiples mutaciones, ya sea que disminuyan de forma directa la afinidad del fármaco por su molécula blanco (mutación primaria) o que se desarrollen para compensar la pérdida de la eficacia biológica del virus (mutación secundaria), que se observa comúnmente después de la aparición de mutaciones primarias (18).
La resistencia a los medicamentos puede estar presente desde antes de iniciar la TARAA (resistencia primaria) o puede ser adquirida con posterioridad al inicio del tratamiento (resistencia secundaria); la primaria se da principalmente por la infección inicial con una cepa de VIH que ya es resistente a uno o más antirretrovirales. En Estados Unidos y Europa recientes estudios sugieren que el riesgo de transmitir un virus resistente a uno o más medicamentos antirretroviral está entre el 6 y 16 %, aunque generalmente menos del 5 % de los virus transmitidos exhiben resistencia a más una clase de ARV (19). En Colombia las frecuencias de resistencia primaria reportadas hasta ahora oscilan entre el 5,8 % y 11,8 % (20, 21).
CLASES DE ANTIRRETROVIRALES Y MECANISMOS DE RESISTENCIA
Los medicamentos antirretrovirales se dividen en seis clases: inhibidores nucleósidos o nucleótidos de transcriptasa reversa (INTR), inhibidores no nucleósidos de transcriptasa reversa (INNTR), inhibidores de proteasa (IP), inhibidores de fusión, bloqueadores del correceptor CCR5 e inhibidores de integrasa (INI) (22). Para 2013, se encontraban 27 antirretrovirales aprobados por la Agencia de Drogas y Alimentos de Estados Unidos (FDA, por sus siglas en inglés), los cuales se han utilizado en el diseño de esquemas de tratamiento contra el VIH (18).
Inhibidores nucleósidos/nucleótidos de transcriptasa reversa (INTR)
Son administrados como pro-medicamentos, es decir, moléculas que requieren ser fosforiladas por quinasas celulares para convertirse en nucleótidos trifosfato, la forma activa del fármaco (23); son análogos de los nucleósidos modificados en el carbono 5' de la desoxirribosa. Se incorporan a la cadena de ADN en formación y previenen la formación del enlace con el próximo nucleótido, resultando en la terminación de la elongación del ADN proviral (22).
Se han identificado dos mecanismos de resistencia: por mutaciones que permiten que la transcriptasa reversa (TR) discrimine entre un INTR y un dNTP producido naturalmente por la célula, previniendo así que los INTR sean incorporados en la cadena creciente de ADN viral; y por mutaciones que median la escisión dependiente de ATP del INTR que ya se encuentra incorporado a la cadena creciente de ADN, permitiendo que la enzima reinicie la síntesis del ADN proviral. Estas mutaciones se conocen como mutaciones de análogos de timidina (TAM, por su nombre en inglés) pues son seleccionadas por análogos de timidina como zidovudina y estavudina (24).
Inhibidores no nucleósidos de transcriptasa reversa (INNTR)
Se unen a la transcriptasa reversa induciendo un cambio conformacional en algunos residuos importantes y hacen más rígida la estructura de dicha enzima, bloqueando la síntesis del ADN. A diferencia de los primeros, estos no requieren del metabolismo celular para ejercer su actividad (25). Regularmente, las mutaciones que se asocian con resistencia a estos medicamentos afectan de manera específica las unión entre el inhibidor y la transcriptasa reversa (26).
Inhibidores de proteasa (IP)
La inhibición de la actividad de la proteasa evita que se dé el procesamiento de las poliproteínas Gag y Gag-Pol, necesario para convertir las nuevas partículas virales nacientes inmaduras no infecciosas, en partículas virales maduras e infecciosas (27). Se dividen en dos grupos: mutaciones primarias o mayores y mutaciones compensatorias
o menores (28).
Las primarias son aquellas que afectan directamente el sitio de unión del inhibidor de proteasa a la enzima; aquí se encuentran, tanto las que se dan en el sitio activo de la proteasa, como las localizadas en regiones cercanas al sitio de corte, las poliproteínas Gag y Gag-Pol.
Las mutaciones menores o compensatorias generalmente son seleccionadas después de tratamiento con los inhibidores de proteasa y se presentan en codones que codifican aminoácidos fuera del sitio activo de la enzima, permitiendo que el virus recupere la eficacia biológica que pudo haber sido comprometida por las mutaciones mayores, contribuyendo indirectamente con la resistencia a los IP (28).
Inhibidores de entrada
La entrada del VIH a la célula no solo está mediada por la unión de la glicoproteína viral gp120 con el receptor celular CD4, es necesario que se genere una segunda interacción con un correceptor celular bien sea CCR5 o CXCR4. La elección del correceptor usado por el virus, es decir, el tropismo viral, está estrechamente relacionado con la adquisición y patogénesis de la enfermedad. En general, la mayoría de los virus transmitidos utilizan el correceptor CCR5 para entrar a las células, mientras que la aparición de virus con tropismo CXCR4 se ha asociado a una mayor velocidad de progresión a SIDA (29).
Los inhibidores de entrada pueden subdividirse en dos clases: inhibidores de fusión y bloqueadores del correceptor CCR5. Los inhibidores de fusión se unen al dominio HR1 y HR2 de la glicoproteína viral gp41, generando un impedimento estérico, que impide la fusión de la envoltura viral con la membrana celular. La resistencia a estos fármacos se genera por mutaciones en HR1 y HR2.
Los bloqueadores del correceptor interactúan con la molécula CCR5, generando un cambio conformacional que evita la unión con la gp120 viral (18). La resistencia a los bloqueadores de CCR5 se da a través del uso de un correceptor alterno CXCR4 debido al desarrollo de mutaciones en gp120 o por mutaciones en gp 41 que permiten eludir el bloqueo de la interacción entre estas glicoproteínas virales y el CCR5 celular (30).
Inhibidores de integrasa (INI)
Son moléculas estructuralmente diversas que contienen un motivo de unión a un catión metálico divalente como Mg2+ o Mn2+ y una región hidrofóbica para entrar en la cavidad formada por la integrasa y el extremo 3' del ADN viral que contiene el dinucleótido terminal CA (31).
La resistencia a inhibidor de integrasa es causada por mutaciones primarias y secundarias a través de tres mecanismos diferentes: el primero es mediado por mutaciones que generan cambios conformacionales dentro del bolsillo catalítico de la integrasa, lo cual lleva a que el inhibidor de integrasa se una a sitios adyacentes al sitio activo de la integrasa, disminuyendo su función significativamente por una inhibición alostérica parcial (32).
El segundo mecanismo es mediado por mutaciones generadas en respuesta al tratamiento con raltegravir, donde se da un cambio en dos aminoácidos ubicados cerca del sitio catalítico de la enzima que forman un puente de hidrógeno con un residuo ubicado dentro de éste, inhibiendo la unión del cofactor metálico necesario para la acción de la integrasa, lo que disminuye la eficacia biológica del virus. Finalmente, el tercer mecanismo es dirigido por mutaciones que disminuyen la unión entre el raltegravir y la integrasa, interfiriendo directamente con la actividad del inhibidor de integrasa (31).
DINÁMICA POBLACIONAL DE LA RESISTENCIA
Una de las características más importante del virus es su alta variabilidad genética, lo que permite la formación de variantes virales con capacidad para evadir el sistema inmune, cambiar el tropismo celular y desarrollar resistencia a medicamentos antirretrovirales, las cuales pueden permanecer como poblaciones minoritarias o, de acuerdo al ambiente, expandirse e influir en el desenlace clínico y en la respuesta al tratamiento.
La transcriptasa reversa del virus se considera la principal fuente de mutaciones, mientras que la segunda mayor contribución a la diversidad del virus es dada por el intercambio de segmentos o recombinación entre dos genomas ARN durante el proceso de duplicación del genoma (33).
La tasa de mutación se define como el número de bases incorrectas que son incorporadas en el genoma del virus en cada ciclo de replicación; esta varía entre 3,4 x 10-5Â a 2,4 x 10-5Â sustituciones/ nucleótido/ciclo replicativo (34); teniendo en cuenta que el genoma viral es de aproximadamente 104 nucleótidos, se puede asumir que aproximadamente una cuarta parte de los viriones nacientes presentan mutaciones (35).
Durante la transcripción del genoma, la transcriptasa reversa debe hacer dos saltos o cambios de molde para generar el ADN proviral (25). Dado que el genoma del VIH está compuesto por dos copias de ARN de cadena sencilla y que un individuo puede infectarse simultáneamente con dos subtipos o dos variantes diferentes, estos saltos durante la transcripción reversa llevan a la formación de partículas virales con genomas recombinantes que pueden contener varias mutaciones que conducen a multiresistencia (36).
La ocurrencia de tales cambios genéticos no necesariamente se expande a la totalidad de la población viral. En ausencia de un factor de selección -como lo puede ser el tratamiento antiviral- dichos mutantes y recombinantes pueden permanecer como variantes minoritarias. El tratamiento con fármacos antirretrovirales puede seleccionarlos positivamente y llevarlos a constituirse en la subpoblación predominante. A su vez, la suspensión del tratamiento puede reversar dicho efecto y llevar a los mutantes resistentes de nuevo a una proporción minoritaria.
Una sola mutación o una combinación particular de mutaciones pueden conferir resistencia a varios medicamentos de la misma clase, esto es, resistencia cruzada. El número de mutaciones requeridas para conferir resistencia y el costo en la eficacia biológica de estas mutaciones constituyen la llamada “barrera genética a la resistencia”, que, en general, es más alta para los IP y más baja para los INNTR y algunos INTR como lamivudina y emtricitabina(18).
MUTACIONES MÁS COMUNES ASOCIADAS A RESISTENCIA A MEDICAMENTOS ANTIRRETROVIRALES
El estudio genético de los diferentes aislados virales permite identificar las diferentes mutaciones asociadas con resistencia a los medicamentos antiretrovirales, si se identifican en al menos una de las siguientes situaciones (37): i) en experimentos in vitro que confirmen la contribución a la resistencia usando mutagénesis dirigida; ii) por evaluación de la disminución de la susceptibilidad in vitro a los antirretrovirales en aislados clínicos o cepas de laboratorio; y iii) por correlaciones entre la secuencia del genoma viral (genotipo) y la respuesta virológica en pacientes que se expusieron al tratamiento (38).
Así, se han podido catalogar las principales mutaciones que generan resistencia a cada uno de estos fármacos las cuales pueden ser consultadas en las bases de datos de resistencia a antirretroviral como la de la Universidad de Standford (http://hivdb.stanford.edu/), la del Laboratorio Nacional Los Alamos del Departamento de Salud y Servicios Humanos de los Estados Unidos (LANL) (http://www.hiv.lanl.gov/content/ index) y la del Departamento de Salud Nacional del Reino Unido de Resistencia a Medicamentos (http://www.hivrdb.org.uk/).
Resistencia a inhibidores nucleósidos/ nucleótidos de transcriptasa reversa (INTR)
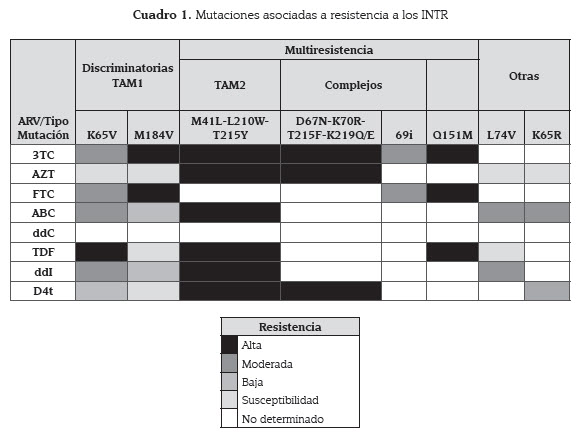
En el cuadro 1 se muestra las principales mutaciones reportadas para las INTR y sus patrones de resistencia. Dentro de las mutaciones discriminatorias, las más comunes son la mutación K65V, en la cual se da un remplazo de lisina (K) por una valina (V) en la posición 65 y la M184V donde se da una cambio de metionina (M) por valina (V) en la posición 184. Sin embargo, también se pueden presentar frecuentemente otras mutaciones como las TAM, las asociadas a resistencia cruzada y otras no clasificadas en los grupos anteriores. A continuación se detallan las características de cada mutación:
* K65V: mutación discriminatoria común ante los inhibidores nucleósidos/nucleótidos de la TR, es seleccionada inicialmente por tenofovir (TDF) y en menor medida por abacavir (ABC), didanosina (ddI), lamivudina (3TC) y emtribicitabina (FTC). Genera una baja resistencia a estavudina (d4T) pero incrementa la susceptibilidad a zidovudina (AZT).
* M184V: es la mutación discriminatoria más común de resistencia ante los inhibidores nucleósidos/nucleótidos de la TR; in vitro genera resistencia elevada a 3TC y FTC, baja resistencia a ddI y el ABC, y susceptibilidad aumentada a la AZT, d4T y al TDF (39).
* Mutaciones de resistencia cruzada: en respuesta a diferentes inhibidores nucleósidos/ nucleótidos de la TR, se pueden generar mutaciones que confieren resistencia a más de un INTR. La inserción en el codón 69 (69i) genera resistencia a todo el grupo de los INTR, pero sólo resistencia intermedia a 3TC y FTC (40). La mutación Q151M genera resistencia a TDF, 3TC y FTC (41); además esta última mutación, se desarrolla en el 5 % de los pacientes que reciben ddI en combinación con AZT o d4T (42).
Las TAM sólo son seleccionadas con terapias que contienen AZT o d4T pero generan resistencia cruzada a otros INTR y ocurren independientemente de las mutaciones discriminatorias; además, se ha observado que la presencia de TAM predispone al desarrollo de otras mutaciones cuando se administra TDF, ABC y ddI.
Las TAM se dividen en dos patrones diferentes denominados TAM-1 y TAM-2, donde para el TAM-1 se observan las mutaciones M41L, L210W y T215Y que generan una resistencia alta a análogos de timidina y resistencia cruzada a ABC, ddI y TDF, mientras que el TAM-2 incluye las mutaciones D67N, K70R, T215F, y K219Q/E (43).
• Otras mutaciones: L74V y K65R se generan en ausencia de análogos de timidina. La K65R causa resistencia intermedia a d4T y susceptibilidad incrementada a AZT. La L74V causa resistencia intermedia a ddI y ABC, y un leve aumento de susceptibilidad a AZT y TDF (37).
Resistencia a inhibidores no nucleósidos de transcriptasa reversa (INNTR)
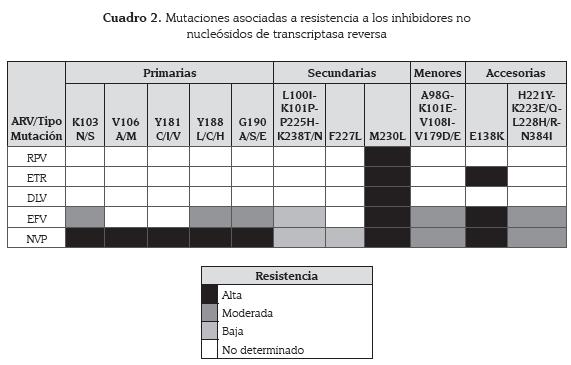
Las mutaciones de resistencia más comunes para los INNTR se clasifican en cuatro categorías diferentes (cuadro 2).
• Primarias: aquellas que causan resistencia a uno o más INNTR y son las primeras que se desarrollan dentro de las terapias con estos medicamentos; entre éstas se encuentran las mutaciones K103N/S, V106A/M, Y181C/I/V, Y188L/C/H y G190A/S/E que generan alta resistencia a la neviparina. Con excepción de las mutaciones V106A and Y181CIV, las demás mutaciones causan alta resistencia o resistencia intermedia al efavirenz (44).
• Secundarias: usualmente se desarrollan en combinación con una mutación primaria y tienen implicaciones en la selección de medicamentos para su uso; entre éstas se encuentran L100I, K101P, P225H, F227L, M230L y K238T/N.
Las mutaciones L100I y K101P ocurren en combinación con K103N y disminuyen la susceptibilidad a NVP y EFV hasta 80 veces en comparación con solo la presencia de K103N (45); para P225H y K238T/N, las cuales usualmente se presentan en combinación con K103N, se observa un efecto sinérgico en la disminución de susceptibilidad a NVP y EFV (46)7F. En el caso de F227L, ésta se presenta en combinación con V106A y dirigen la reducción sinérgica de susceptibilidad a neviparina y, finalmente, M230L puede presentarse sola y generar una disminución en la susceptibilidad hasta 20 veces más a todos los INNTR incluyendo la etravirina (ETV) (46).
• Mutaciones menores no polimórficas: pueden ocurrir solas o en combinación con otras mutaciones asociadas a resistencia y que causan un reducción baja pero consistente en la susceptibilidad a los inhibidores no nucleósidos de la TR. Las mutaciones de este grupo son A98G, K101E, V108I y reducen la susceptibilidad a neviparina y efavirenz. V179D unida a K103R que per se no genera resistencia, disminuye la susceptibilidad a neviparina y efavirenz (47)/.
• Mutaciones accesorias polimórficas: modulan el efecto de otras mutaciones de resistencia a los inhibidores no nucleósidos de la TR; un ejemplo de esto es la mutación E138K, que parece conferir resistencia cruzada a etravirina, neviparina y efavirenz. Las mutaciones K101P/ H/E, I135T/M y V179D/E/F/T/L disminuyen la susceptibilidad a neviparina y efavirenz (42).
Resistencia a inhibidores de proteasa (IP)
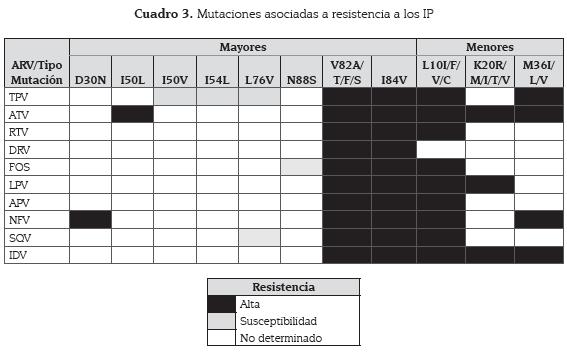
Las mutaciones de resistencia pueden ser (27):
• Mutaciones mayores o primarias: por sí solas generan una reducción en la susceptibilidad para uno o más inhibidores de proteasa. Las más comunes son D30N, V32I, M46IL, G48V/M, I50V/L, I54V/T/A/L/M, L76V, V82A/T/F/S, I84V, N88S y L90M (18). D30N es seleccionada por nelfinavir (NFV) y I50L por atazanavir (ATV), siendo las únicas mutaciones que generan resistencia a un solo inhibidor de proteasa (48); además, se ha observado que la mutación D30N ocurre con mayor frecuencia en virus subtipo B y L90M en virus subtipo C, F, G y CRF01_AE (49).
Las mutaciones I50V e I54L incrementan la susceptibilidad a tripanavir (TPV), N88S incrementa la susceptibilidad a fosamprenavir y L76V incrementa la susceptibilidad a atazanavir, saquinavir y tripanavir (42); mientras que V82A/T/F/S y I84V están asociados a resistencia a todos los inhibidores de proteasa, resaltando por ser mutaciones con un alto grado de polimorfismo (37).
• Mutaciones compensatorias o menores: se encargan de compensar las alteraciones en la eficiencia biológica del virus originada por mutaciones mayores, pueden ser polimorfismos prexistentes o adquiridos después de las mutaciones primarias y se han encontrado en aislados de pacientes sin tratamiento. Las mutaciones accesorias que regulan positivamente la función de la proteasa compensan las mutaciones mayores y se ubican en las posiciones 10, 20, 36 y 71 de esta enzima viral. Las posiciones 10, 20 y 36 son altamente polimórficas, presentándose asociaciones de las mutaciones en la posición 10 con resistencia la mayoría de los IP, excepto para el darunavir. Las mutaciones en la posición 20 están asociadas a resistencia a indinavir, lopinavir y atazanavir, mientras que las mutaciones en la posición 36 generan resistencia a atazanavir, indinavir, nelfinavir y tripanavir. Además, se han reportado otras mutaciones polimórficas como I13V, D60E, I62V, V77I y I93L, y otras mutaciones no polimórficas no comunes que incluyen V11I, E34Q, E35G, K43T, K45I, K55R, Q58E, T74P/A/S, V75I, N83D, P79A/S, I85V, L89V, T91S, Q92K y C95F (cuadro 3) (50).
• Combinación de mutaciones mayores y accesorias: se han observado en la mayoría de los virus resistentes a los inhibidores de proteasa.
Resistencia a los inhibidores de integrasa (INI)
A la fecha, dos INI han sido aprobados por la FDA: raltegravir y el dolutegravir. Para raltegravir se distinguen tres patrones de mutaciones conformados por una mutación mayor (N155H, Q148H/R/K y Y143C/R) y una mutación menor (E92Q, G140A/S y T97A). El primer patrón se distingue por la presencia de las mutaciones N155H y E92Q; el segundo por las mutaciones Q148H/R/K y G140S/A y el tercero por las mutaciones Y143C/R y T97A (32), a pesar de la reciente aprobación del dolutegravir en 2013, las mutaciones Q148H y G140S en combinación con las mutaciones L74I/M, E92Q, T97A,E138A/K, G140A, o N155H se han asociado con una susceptibilidad reducida entre 5 -20 veces a este medicamento, además de una reducción en la supresión viral (51).
Resistencia a inhibidores de entrada
Para el enfuvirtide (ENF), el único inhibidor de fusión aprobado por la FDA, se reconocen las mutaciones desde el residuo 36 hasta el 45 en el gen de la envoltura, siendo las mutaciones G36D/E/V, V38E/A, Q40H, N42T y N43D las más importantes.
Además se reconocen tres mutaciones accesorias (N126K, N137K y S138A) que se asocian con una reconstitución de la eficacia biológica (37). En el caso de los bloqueadores del correceptor CCR5, solo se ha aprobado el maraviroc; ya se han identificado mutaciones en el asa V3 de la gp120 en pacientes que incluyen este fármaco en su esquema de tratamiento, pero además también se han reportado la presencia de mutaciones en gp41 sin mutaciones en el asa V3, por lo cual aún no se ha establecido el papel de estas mutaciones en la inducción de resistencia ni su significancia clínica, ya que se ha observado que se genera resistencia sin cambiar el tropismo del virus (37).
EVALUACIÓN DE LA RESISTENCIA A ANTIRRETROVIRALES
Actualmente se encuentran disponibles en el mercado varias técnicas para evaluar resistencia a antirretrovirales, las cuales se clasifican en pruebas fenotípicas y pruebas genotípicas. Las pruebas fenotípicas buscan establecer la concentración necesaria de un fármaco para inhibir en un 50 % (IC50) la replicación de la cepa viral en estudio (52).
Aunque este es el método más directo de detectar y cuantificar la resistencia a los diferentes fármacos antirretrovirales, estas pruebas presentan ciertas desventajas: no pueden realizarse en individuos que tienen cargas virales bajas, el análisis se debe hacer independientemente para cada fármaco, el procedimiento demora varios días y su ejecución presenta elevados costos. Sumado a lo anterior, estos tipo de pruebas solamente están disponibles en laboratorios especializados de alta complejidad con capacidad para cultivar el VIH y no en los laboratorios clínicos generales (53).
Por su parte, las pruebas genotípicas emplean análisis de secuencias del genoma viral para detectar la presencia de las mutaciones asociadas con resistencia a antiretrovirales. Este tipo de pruebas, las más usadas en los laboratorios de diagnóstico, utilizan diferentes herramientas bioinformáticas para el análisis in sílico de las secuencias. La técnica permite identificar mutaciones asociadas a resistencia presentes en la cepa infectante. Entre las ventajas de este método se destaca el corto tiempo entre la toma de muestra y la obtención de un resultado y la capacidad de predecir la resistencia antes de que aparezca el fenotipo resistente.
Entre sus limitaciones se encuentra el que estas tampoco pueden realizarse en individuos que tienen cargas virales bajas y es posible que no sean detectadas las variantes minoritarias resistentes que representen menos del 20% del total de la población viral. Además, en el momento de la obtención de la muestra se debe continuar con los antirretrovirales para mantener la presión selectiva y así evitar que las variantes minoritarias vuelvan a una pequeña proporción (54). Aunque aún no está del todo claro el impacto de estas mutantes minoritarias frente a la TARAA, se ha encontrado una asociación entre su presencia y pobres desenlaces clínicos (53).
Se encuentran disponibles programas bioinformáticos gratuitos diseñados específicamente para predecir el subtipo o para identificar mutaciones asociadas a resistencia. Estos permiten además una aproximación cuantitativa al grado de resistencia asociado a cada combinación de mutaciones, mediante la comparación con cepas previamente secuenciadas y evaluadas por métodos fenotípicos; esto constituye el llamado "fenotipo virtual".
Entre estos programas podemos citar Geno2pheno Resistance (http://www.geno2pheno.org/) el cual detecta resistencia genotípica para 15 diferentes medicamentos ARV y permite predecir el subtipo viral (55).
INDICACIONES E IMPLICACIONES CLÍNICAS DE LA EVALUACIÓN DE RESISTENCIA A ANTIRRETROVIRALES
En principio la detección de las diferentes mutaciones es ideal antes del inicio de la terapia antirretroviral altamente activa, ya que permitiría personalizar el esquema de tratamiento. Sin embargo, las indicaciones de las pruebas de resistencia a los antirretrovirales han sido objeto de cierto debate. Para el 2013 los lineamientos en la guía de tratamiento propuesta por el Departamento de Salud de los Estados Unidos (DHHS) sugieren realizar una prueba genotípica después de la primera falla virológica con el fin de seleccionar un nuevo régimen; además se recomienda una evaluación genotípica del tropismo viral por el correceptor, antes de iniciar un esquema de tratamiento que incluya un bloqueador de CCR5 (11).
Por otra parte, la guía de tratamiento de la Sociedad Europea Clínica de SIDA (EACS, según sus siglas en inglés) de 2013 sugiere realizar la prueba genotípica de resistencia desde antes de iniciar la terapia antirretroviral altamente activa o cuando existe el riesgo de una superinfección con varias cepas de VIH (56). En Colombia no existe una guía actualizada para el manejo de pacientes infectados con VIH; la guía para el manejo del VIH/SIDA basada en la evidencia de 2006, sugería realizar la evaluación genotípica después del segundo o tercer fracaso terapéutico y no realizarla en pacientes con problemas de adherencia o intolerancia a los antirretrovirales (57).
Teniendo en cuenta las sugerencias previamente mencionadas, es previsible que las nuevas guías colombianas -actualmente en proceso de revisión- recomienden la realización de las pruebas genotípicas de resistencia previamente al inicio de la TARAA, o al menos después de la primera falla, con lo cual probablemente se podría aumentar la probabilidad del éxito terapéutico así como la expectativa y la calidad de vida de los pacientes.
CONCLUSIONES
Una de las principales características del VIH es su diversidad genética, que se genera, en parte, por la acumulación de mutaciones insertadas al azar por la transcriptasa reversa, las cuales pueden inducir no solo escape inmunológico sino también resistencia a los fármacos antirretrovirales, lo que se traduce en falla virológica.
Teniendo en cuenta que diferentes estudios han demostrado que la progresión de la enfermedad puede ser modulada por el inicio temprano de la terapia antirretroviral altamente activa, se hace necesario diseñar tanto políticas públicas como guías clínicas que evalúen detalladamente las características clínicas de cada individuo infectado, con el fin de establecer el esquema más adecuado y reducir al máximo la aparición y/o selección de variantes virales resistentes al tratamiento.
En Colombia existe una guía desde el año 2006, donde se describe el protocolo de clasificación y manejo de los pacientes VIH; sin embargo, desde esa fecha no se han realizado actualizaciones, por lo que se continúa evaluando el perfil de resistencia por pruebas genotípicas sólo después de la segunda o tercera falla virológica, paradigma que debe ser re-evaluado, dado que este comportamiento afecta la respuesta a la TARAA y promueve la aparición de mutaciones asociadas a resistencia.
Es importante realizar estudios descriptivos que identifiquen los medicamentos y esquemas de mayor uso para la terapia antirretroviral altamente activa en Colombia y la frecuencia de fallas virológicas por paciente; esta información ayudaría a monitorear el perfil de resistencia a antirretrovirales en nuestra población.
CONFLICTOS DE INTERESES
Los autores del presente trabajo declaran no presentar conflicto de intereses.
AGRADECIMIENTOS
A Colciencias por la financiación del proyecto Nº. 111556933380.
REFERENCIAS
1. UNAIDS. Global Report United States: United Nations Programme on HIV/AIDS (UNAIDS); 2012 (cited 2013 January). [ Links ]
2. Social MdSPyP. Informe UNGASS - 2012 Seguimiento de la Declaración de compromiso sobre el VIH/Sida. 2012. [ Links ]
3. Namme Luma H, Doualla MS, Choukem SP, Temfack E, Ashuntantang G, Achu Joko H, et al. Adverse drug reactions of Highly Active Antiretroviral Therapy (HAART) in HIV infected patients at the General Hospital, Douala, Cameroon: a cross sectional study. Pan Afr Med J. 2012; 12:87. [ Links ]
4. Swanstrom R, Coffin J. HIV-1 pathogenesis: the virus. Cold Spring Harb Perspect Med. 2012; 2(12):a007443. [ Links ]
5. Lederman MM, Connick E, Landay A, Kuritzkes DR, Spritzler J, St Clair M, et al. Immunologic responses associated with 12 weeks of combination antiretroviral therapy consisting of zidovudine, lamivudine, and ritonavir: results of AIDS Clinical Trials Group Protocol 315. J Infect Dis. 1998; 178(1):70-9. [ Links ]
6. UNAIDS. Regional Fact Sheet 2012 - Latin America And The Caribbean United States 2012 (cited 2013). [ Links ]
7. Kitahata MM. When to start antiretroviral therapy. Top HIV Med. 2010; 18(3):121-6. [ Links ]
8. Mills FP, Ford N, Nachega JB, Bansback N, Nosyk B, Yaya S, et al. Earlier initialization of highly active antiretroviral therapy is associated with long-term survival and is costeffective: findings from a deterministic model of a 10-year Ugandan cohort. J Acquir Immune Defic Syndr. 2012; 61(3):364-9. [ Links ]
9. Ghaffari G, Passalacqua DJ, Caicedo JL, Goodenow MM, Sleasman JW. Two-year clinical and immune outcomes in human immunodeficiency virus-infected children who reconstitute CD4 T cells without control of viral replication after combination antiretroviral therapy. Pediatrics. 2004; 114(5):e604-11. [ Links ]
10. Kuller LH, Tracy R, Belloso W, De Wit S, Drummond F, Lane HC, et al. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med. 2008; 5(10):e203. [ Links ]
11. Antiretroviral DoHaHSPo. Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. 2013. p. 3:4. [ Links ]
12. Wainberg MA. Combination therapies, effectiveness, and adherence in patients with HIV infection: clinical utility of a single tablet of emtricitabine, rilpivirine, and tenofovir. HIV AIDS (Auckl). 2013;5:41-9. [ Links ]
13. von Wyl V, Klimkait T, Yerly S, Nicca D, Furrer H, Cavassini M, et al. Adherence as a predictor of the development of class-specific resistance mutations: the Swiss HIV Cohort Study. PLoS One. 2013;8(10):e77691. [ Links ]
14. van der Kuyl AC, Cornelissen M. Identifying HIV-1 dual infections. Retrovirology. 2007;4:67. [ Links ]
15. Gottlieb GS, Nickle DC, Jensen MA, Wong KG, Grobler J, Li F, et al. Dual HIV-1 infection associated with rapid disease progression. Lancet. 2004;363(9409):619-22. [ Links ]
16. Wainberg MA, Brenner BG. Role of HIV Subtype Diversity in the development of resistance to antiviral drugs. Viruses. 2010; 2(11):2493-508. [ Links ]
17. Eshleman SH, Hoover DR, Chen S, Hudelson SE, Guay LA, Mwatha A, et al. Nevirapine (NVP) resistance in women with HIV-1 subtype C, compared with subtypes A and D, after the administration of single-dose NVP. J Infect Dis. 2005; 192(1):30-6. [ Links ]
18. Tang MW, Shafer RW. HIV-1 antiretroviral resistance: scientific principles and clinical applications. Drugs. 2012; 72(9):e1-25. [ Links ]
19. Cohen MS, Chen YQ, McCauley M, Gamble T, Hosseinipour MC, Kumarasamy N, et al. Prevention of HIV-1 infection with early antiretroviral therapy. N Engl J Med. 2011; 365(6):493-505. [ Links ]
20. Galindo-Orrego P, Mueses-Marín HF, Galindo- Quintero J, Martínez-Cajas JL. Resistencia transmitida del virus de la inmunodeficiencia humana en pacientes sin exposición previa a tratamiento antirretroviral, Cali, Colombia, 2010. Infectio. 2014; 17:19-27. [ Links ]
21. DiazGranados CA, Mantilla M, Lenis W. Antiretroviral drug resistance in HIV-infected patients in Colombia. Int J Infect Dis. 2010; 14(4):e298-303. [ Links ]
22. Arts EJ, Hazuda DJ. HIV-1 Antiretroviral Drug Therapy. Cold Spring Harb Perspect Med. 2012; 2(4):a007161. [ Links ]
23. Nakashima H, Matsui T, Harada S, Kobayashi N, Matsuda A, Ueda T, et al. Inhibition of replication and cytopathic effect of human T cell lymphotropic virus type III/lymphadenopathy- associated virus by 3'-azido-3'- deoxythymidine in vitro. Antimicrob Agents Chemother. 1986;30(6):933-7. [ Links ]
24. Clavel F, Hance AJ. HIV drug resistance. N Engl J Med. 2004; 350(10):1023-35. [ Links ]
25. Esposito F, Corona A, Tramontano E. HIV- 1 Reverse transcriptase still remains a new drug target: structure, function, classical inhibitors, and new inhibitors with innovative mechanisms of actions. Mol Biol Int. 2012; 2012:586401. [ Links ]
26. Maga G, Amacker M, Ruel N, Hübscher U, Spadari S. Resistance to nevirapine of HIV-1 reverse transcriptase mutants: loss of stabilizing interactions and thermodynamic or steric barriers are induced by different single amino acid substitutions. J Mol Biol. 1997; 274(5):738-47. [ Links ]
27. Adamson CS. Protease-Mediated Maturation of HIV: Inhibitors of protease and the maturation process. Mol Biol Int. 2012;2012:604261. [ Links ]
28. Henderson GJ, Lee SK, Irlbeck DM, Harris J, Kline M, Pollom E, et al. Interplay between single resistance-associated mutations in the HIV-1 protease and viral infectivity, protease activity, and inhibitor sensitivity. Antimicrob Agents Chemother. 2012; 56(2):623-33. [ Links ]
29. Didigu CA, Doms RW. Novel approaches to inhibit HIV entry. Viruses. 2012; 4(2):309-24. [ Links ]
30. Gilliam BL, Riedel DJ, Redfield RR. Clinical use of CCR5 inhibitors in HIV and beyond. J Transl Med. 2011; 9 Suppl 1:S9. [ Links ]
31. Blanco JL, Varghese V, Rhee SY, Gatell JM, Shafer RW. HIV-1 integrase inhibitor resistance and its clinical implications. J Infect Dis. 2011; 203(9):1204-14. [ Links ]
32. Quashie PK, Mesplede T, Wainberg MA. HIV Drug resistance and the advent of integrase inhibitors. Curr Infect Dis Rep. 2013; 15(1):85-100. [ Links ]
33. Chen J, Powell D, Hu WS. High frequency of genetic recombination is a common feature of primate lentivirus replication. J Virol. 2006; 80(19):9651-8. [ Links ]
34. Mansky LM, Temin HM. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J Virol. 1995; 69(8):5087-94. [ Links ]
35. Simon V, Ho DD. HIV-1 dynamics in vivo: implications for therapy. Nat Rev Microbiol. 2003; 1(3):181-90. [ Links ]
36. Mougel M, Houzet L, Darlix JL. When is it time for reverse transcription to start and go? Retrovirology. 2009; 6:24. [ Links ]
37. Johnson VA, Calvez V, Günthard HF, Paredes R, Pillay D, Shafer R, et al. 2011 update of the drug resistance mutations in HIV-1. Top Antivir Med. 2011;19(4):156-64. [ Links ]
38. Database SHDR. Release Notes for HIVdb, HIVsew, HIValg 2012 (cited 2013). Available from: http://hivdb.stanford.edu/DR/asi/releaseNotes/index.html#hivdb_mutationcategories. [ Links ]
39. Whitcomb JM, Parkin NT, Chappey C, Hellmann NS, Petropoulos CJ. Broad nucleoside reversetranscriptase inhibitor cross-resistance in human immunodeficiency virus type 1 clinical isolates. J Infect Dis. 2003; 188(7):992-1000. [ Links ]
40. Prado JG, Franco S, Matamoros T, Ruiz L, Clotet B, Menéndez-Arias L, et al. Relative replication fitness of multi-nucleoside analogueresistant HIV-1 strains bearing a dipeptide insertion in the fingers subdomain of the reverse transcriptase and mutations at codons 67 and 215. Virology. 2004; 326(1):103-12. [ Links ]
41. Shirasaka T, Kavlick MF, Ueno T, Gao WY, Kojima E, Alcaide ML, et al. Emergence of human immunodeficiency virus type 1 variants with resistance to multiple dideoxynucleosides inpatients receiving therapy with dideoxynucleosides. Proc Natl Acad Sci U S A. 1995; 92(6):2398-402. [ Links ]
42. Shafer RW, Schapiro JM. HIV-1 drug resistance mutations: an updated framework for the second decade of HAART. AIDS Rev. 2008; 10(2):67-84. [ Links ]
43. De Luca A, Giambenedetto SD, Trotta MP, Colafigli M, Prosperi M, Ruiz L, et al. Improved interpretation of genotypic changes in the HIV-1 reverse transcriptase coding region that determine the virological response to didanosine. J Infect Dis. 2007; 196(11):1645-53. [ Links ]
44. Rhee SY, Taylor J, Wadhera G, Ben-Hur A, Brutlag DL, Shafer RW. Genotypic predictors of human immunodeficiency virus type 1 drug resistance. Proc Natl Acad Sci U S A. 2006; 103(46):17355-60. [ Links ]
45. Bacheler L, Jeffrey S, Hanna G, D'Aquila R, Wallace L, Logue K, et al. Genotypic correlates of phenotypic resistance to efavirenz in virus isolates from patients failing nonnucleoside reverse transcriptase inhibitor therapy. J Virol. 2001; 75(11):4999-5008. [ Links ]
46. Vingerhoets J, Azijn H, Fransen E, De Baere I, Smeulders L, Jochmans D, et al. TMC125 displays a high genetic barrier to the development of resistance: evidence from in vitro selection experiments. J Virol. 2005; 79(20):12773-82. [ Links ]
47. Rhee SY, Gonzales MJ, Kantor R, Betts BJ, Ravela J, Shafer RW. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res. 2003; 31(1):298-303. [ Links ]
48. Rhee SY, Taylor J, Fessel WJ, Kaufman D, Towner W, Troia P, et al. HIV-1 protease mutations and protease inhibitor cross-resistance. Antimicrob Agents Chemother. 2010; 54(10):4253-61. [ Links ]
49. Calazans A, Brindeiro R, Brindeiro P, Verli H, Arruda MB, Gonzalez LM, et al. Low accumulation of L90M in protease from subtype F HIV-1 with resistance to protease inhibitors is caused by the L89M polymorphism. J Infect Dis. 2005; 191(11):1961-70. [ Links ]
50. Rhee SY, Fessel WJ, Zolopa AR, Hurley L, Liu T, Taylor J, et al. HIV-1 Protease and reversetranscriptase mutations: correlations with antiretroviral therapy in subtype B isolates and implications for drug-resistance surveillance. J Infect Dis. 2005; 192(3):456-65. [ Links ]
51. Johnson VA, Calvez V, Gunthard HF, Paredes R, Pillay D, Shafer RW, et al. Update of the drug resistance mutations in HIV-1: March 2013. Top Antivir Med. 2013; 21(1):6-14. [ Links ]
52. Pillay D, Zambon M. Antiviral drug resistance. BMJ. 1998; 317(7159):660-2. [ Links ]
53. García F, álvarez M, Bernal C, Chueca N, Guillot V. Laboratory diagnosis of HIV infection, viral tropism and resistance to antiretrovirals. Enferm Infecc Microbiol Clin. 2011; 29(4):297-307. [ Links ]
54. Li JZ, Paredes R, Ribaudo HJ, Kozal MJ, Svarovskaia ES, Johnson JA, et al. Impact of minority nonnucleoside reverse transcriptase inhibitor resistance mutations on resistance genotype after virologic failure. J Infect Dis. 2013;207(6):893-7. [ Links ]
55. Beerenwinkel N, Däumer M, Oette M, Korn K, Hoffmann D, Kaiser R, et al. Geno2pheno: Estimating phenotypic drug resistance from HIV-1 genotypes. Nucleic Acids Res. 2003; 31(13):3850-5. [ Links ]
56. Society EAC. European Guidelines for treatment of HIV-infected adults in Europe. 7.0 ed2013. [ Links ]
57. DíazGranados C, álvarez C, Prada G, Martínez F, Sarmiento C. Guía para el manejo de vih/ sida basada en la evidencia Colombia. 2006. [ Links ]
Recibido en: agosto 26 de 2013. Revisado en: marzo 6 de 2014. Aceptado en: marzo 13 de 2014.
