










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkCES Medicina
versão impressa ISSN 0120-8705
CES Med. vol.29 no.1 Medellín jan./jun. 2015
REVISIÓN DE TEMA
Mecanismos de señalización por β-catenina y su papel en la carcinogénesis
β-catenin signaling mechanisms and its role in carcinogenesis
CAROLINA MANTILLA,1 IRIS SUÁREZ MELLADO,2 ALEJANDRA DUQUE JARAMILLO,3 MARIA CRISTINA NAVAS4
1 Bióloga, Grupo de Gastrohepatología. Universidad de Antioquia, Medellín, Colombia.
2 2 Bióloga, MSc Ciencias Básicas, Grupo de Gastrohepatología. Universidad de Antioquia, Medellín, Colombia.
3 Bióloga, joven investigadora, Grupo de Gastrohepatología, Universidad de Antioquia, Medellín, Colombia.
4 MSc, PhD Virología. Profesora Asociada, Departamento de Microbiología y Parasitología, Facultad de Medicina, Coordinadora Grupo Gastrohepatología, Universidad de Antioquia. Medellín, Colombia. maria.navas@udea.edu.co
RESUMEN
La alteración de las vías de señalización es un mecanismo común en la oncogénesis de diferentes tipos de tumor. La modificación de una de las proteínas de la vía por mutaciones o por modificaciones genéticas o epigenéticas en el promotor del gen correspondiente podría generar una alteración en la vía, y por tanto condiciones para el crecimiento descontrolado característico del cáncer.
La vía de señalización Wnt/β-catenina, donde β-catenina actúa como coactivador, es muy importante en procesos de embriogénesis, organogénesis y homeostasis. La alteración de Wnt/β-catenina por mutaciones o modificaciones epigenéticas de β-catenina o de otras proteínas facilita la acumulación de β-catenina en el núcleo y genera una activación permanente de esta vía de señalización. Este evento desencadena la expresión de genes que codifican proteínas que participan en proliferación celular, diferenciación y mantenimiento de células madre.
β-catenina participa también en la adhesión célulacélula, mediando la interacción entre cadherinas y actina. Alteraciones en el complejo β-catenina-cadherina-actina lleva a la pérdida de la adhesión y a una alta capacidad invasiva de las células afectadas, mecanismos asociados con la capacidad de metástasis de las células tumorales.
En esta revisión se describe la proteína β-catenina y su papel en la vía Wnt/ β-catenina, así como en la regulación de la expresión génica y en el proceso de adhesión célula-célula, y las alteraciones que pueden desencadenar un proceso oncogénico.
PALABRAS CLAVE
β-catenina, CTNNB1, Adhesión celular, Wnt/β-catenina, Carcinogénesis.
ABSTRACT
The modification of signaling pathways is a common mechanism in the oncogénesis process of different types of tumor. An alteration in any protein of the pathway by genetic mutations or by epigenetic changes in its gene promoter could cause misregulation of the pathway and therefore lead to uncontrolled cell proliferation.
The Wnt/β-catenin signaling pathway is important in several processes, such as embryogenesis, organogenesis and homeostasis. β-catenin protein acts as a co-activator of this pathway, and when it is translocated to the nucleus, it functions as a transcription factor. An imbalance in this pathway by mutations or epigenetic modifications of β-catenin and/or other proteins, favors the accumulation of β-catenin in the nucleus, leading to permanent activation of Wnt/β-catenin signaling pathway. This event triggers the expression of genes encoding proteins involved in cell proliferation, differentiation and maintenance of stem cells.
β-catenin is also known to participate in cell-cell adhesion, mediating the interaction between Cadherin and Actin. Alterations in the complex β-catenin-Cadherin-Actin lead to a loss of adhesion and a high invasive capacity of affected cells, mechanisms associated with invasiveness of tumor cells. This review describes the β-catenin protein, and its role in the Wnt/β-catenin signaling pathway, as well as in gene expression regulation and cellcell adhesion, and the alterations that can trigger an oncogenic process.
KEY WORDS
β-catenin, CTNNB1, Cell adhesion, Wnt/β-catenin, Carcinogenesis.
INTRODUCCIÓN
La vía de señalización Wnt/β-catenina regula procesos como la regeneración de tejidos, la diferenciación de células madre y la proliferación celular. La alteración de esta vía ha sido descrita en diversos tipos de tumor como el cáncer de colón, cáncer primario de hígado, cáncer de ovario y melanoma, entre otros.
La proteína β-catenina, codificada por el gen CTNNB1, es el principal componente de la vía Wnt/β-catenina. La regulación de esta vía de señalización está basada en la presencia del ligando Wnt que produce la activación; cuando la vía está activa se impide la degradación de laproteína β-catenina por acción de un complejo de proteínas conocido como complejo de destrucción, conformado por quinasas, la proteína adenomatosis poliposis Coli (APC) y la proteína axina. En ausencia del ligando Wnt la vía no está activa y, como consecuencia, la proteína β-catenina es fosforilada y posteriormente degradada en el proteasoma, impidiendo sus funciones como factor de transcripción y en la interacción célula-célula.
Esta proteína participa además en la adhesión celular mediada por cadherina, cuya desregulación se ha asociado también con procesos oncogénicos debido a la pérdida de la adhesión celular y al aumento de la capacidad de invasión de las células tumorales.
Teniendo en cuenta la importancia de esta vía de señalización en procesos como regeneración de tejidos, diferenciación de células madre y proliferación celular, se realizó una revisión de tema sobre los mecanismos de señalización y adhesión célula-célula mediados por β-catenina y su papel en la carcinogénesis. Para esto, se efectuó una búsqueda de literatura de trabajos originales y revisiones publicados entre 1992 y 2014 en la base de datos PubMed. La búsqueda se realizó utilizando las palabras clave: Wnt/β-catenin, signaling pathway, β-catenin, cancer. Los autores escogieron los artículos más relevantes según su criterio para ser incluidos en esta revisión.
En esta revisión se describe la vía Wnt/β- catenina, que funciona en respuesta al coactivador transcripcional β-catenina, y su papel en la regulación de la expresión de proteínas claves en la proliferación celular y diferenciación. Además se presentan las evidencias de desregulación de la vía Wnt/ β-catenina en carcinogénesis, en particular las alteraciones en el gen CTNBB1. Adicionalmente, se describe el papel de β-catenina en la adhesión celular y la regulación de este proceso en cáncer, para finalizar con un recuento de los hallazgos más recientes del papel de los miRNAs sobre las funciones biológicas de la proteína β-catenina.
ESTRUCTURA DE LA PROTEíNA β-CATENINA
β-catenina fue originalmente identificada por su asociación con el dominio citoplasmático de las cadherinas y su papel en la adhesión celular dependiente de Ca 2+. Esta proteína es un componente principal en la adhesión célula-célula por interacción con la proteína cadherina (proteínas de adhesión intercelular); adicionalmente, β-catenina regula la expresión génica a través de la vía de señalización Wnt/β-catenina (1).
β-catenina se encuentra en dos complejos moleculares, que corresponden a estas dos funciones: el primero corresponde a β-catenina como parte de complejos de alto peso molecular implicados en la adhesión celular mediada por E-cadherina; en la segunda función, β-catenina, se encuentra en estado monomérico participando en la señalización de la vía Wnt/β-catenina (2). La coordinación de estas dos vías es fundamental para el control de los procesos como la formación del eje y mesodermo en el embrión, la diferenciación de células madre y en procesos de patogénesis como la carcinogénesis (3).
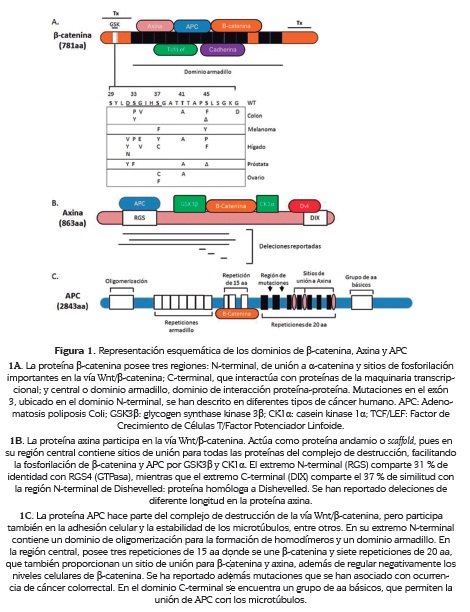
β-catenina es una proteína de 92 kDa, codificada por el gen CTNNB1 que se localiza en el cromosoma 3p22. Esta proteína de 781 aminoácidos consta de tres dominios, amino-terminal, carboxi- terminal y central (4).
El dominio amino-terminal (N-terminal), de aproximadamente 150 aminoácidos, es la región de unión a α-catenina, clave en la interacción célula-célula. En este dominio se localizan los sitios de fosforilación de las quinasas en la vía de señalización Wnt/β-catenina, la glicógeno sintasa quinasa 3β (GSK3β) y la caseína quinasa 1α (CK1α) (5). El dominio carboxi-terminal (C-terminal), de 100 aminoácidos aproximadamente, permite la interacción con proteínas celulares para su papel como factor de transcripción de genes dependientes del factor de crecimiento de células T/ factor potenciador linfoide (Tcf/Lef) (6); genes como Cyclin D1, c-Myc y metaloproteinasas de matriz celular MMP-2, 3, 7, 9, 13.
El dominio central de 550 aminoácidos, está formado por una secuencia de 42 aminoácidos que se repite 12 veces, dominio conocido como "repeticiones armadillo"; esta región que presenta una estructura en hélice, tiene como función la interacción con proteínas como E-cadherina, axina, APC y Tcf/Lef (figura 1). Mediante cristalografía de rayos X se determinó que las 12 repeticiones armadillo forman una superhélice y un surco largo cargado positivamente; este surco interactúa con residuos ácidos consecutivos comunes en cadherinas, APC y miembros de la familia TCF (7).
β-CATENINA EN LA VíA DE SEñALIZACIóN WNT/ β-CATENINA
La vía de señalización Wnt/β-catenina juega un papel crucial en la regeneración de tejidos y en el proceso de diferenciación de células madre (8, 9). Esta vía de señalización fue descrita por primera vez en Drosophila (mosca de la fruta) y fue denominada Wingless (del inglés: "sin alas"), pues alteraciones en esta vía generan un defecto en el desarrollo de las alas y los halterios en la mosca. La vía es indispensable para el correcto patrón celular en el embrión y el desarrollo de los discos imaginales de la larva, constituidos por células indiferenciadas que darán origen a las estructuras cuticulares de la mosca adulta como alas, antenas, entre otras (10).
Wnt-1 es el gen Wnt que más se ha estudiado y corresponde a un proto-oncogén. La primera descripción se realizó en células que presentaban integración del genoma del virus de tumor mamario de ratón (VTMR) cerca del promotor del gen Wnt; la secuencia viral regula este gen celular causando una sobreexpresión del ligando Wnt y por tanto la activación de la vía de señalización (11).
En células de mamíferos la vía de señalización se activa cuando el ligando Wnt se une al receptor Frizzled (Fz) y al receptor de lipoproteínas de baja densidad relacionados con la proteína 5/6 (LRP5/6); la interacción ligandoreceptores produce la fosforilación de LRP5/6 por actividad de las quinasas CK1 y GSK3β, en un dominio conservado proporcionando un sitio óptimo de unión para la proteína axina. La activación de la vía y el reclutamiento de axina y proteínas asociadas (GSK3β, CK1α, APC) a Fz-LRP5/6 en la membrana regulan de forma negativa el denominado complejo de destrucción, conformado por las proteínas GSK3β, CK1α, APC y axina (12, 13). Este complejo tiene como función marcar la proteína β-catenina para que sea degradada en el proteasoma y de este manera impedir su actividad como factor de transcripción.
Si el complejo de destrucción no cumple su función, se produce la acumulación de la forma estable de β-catenina en el citoplasma, la cual es traslocada al núcleo para actuar como factor de transcripción de genes que codifican proteínas involucradas en proliferación celular, diferenciación y mantenimiento de células madre, como c-myc (14), ciclina D (15) y Tcf-1 (16).
La axina proporciona sitios de unión a todas las proteínas del complejo de destrucción y por esto se conoce como proteína "andamio" o scaffold (figura 1B). La fosforilación de axina por GSK3β aumenta la afinidad por las proteínas del complejo, mientras que en su estado hipofosforilado la proteína axina se degrada y se produce la desestabilización del complejo (17, 18). Durante la activación de la vía de señalización, las quinasas se reclutan en un dominio del correceptor LRP5/6 (PPPSPXS) y como consecuencia el complejo se desestabiliza.
Las quinasas GSK3β y CK1α son las encargadas de fosforilar la proteína β-catenina en cuatro residuos del dominio N-terminal. Inicialmente CK1α fosforila el residuo Ser45 y luego GSK3β fosforila los residuos Th41, Ser37 y Ser33 (figura 2) (1, 19). Esta modificación es indispensable para la poliubiquitinación y ulterior degradación de β-catenina en el proteasoma. Como se mencionó anteriormente, si la proteína β-catenina se degrada, no es traslocada al núcleo y por ende no puede cumplir su función como factor de transcripción (figura 2 - vía inactiva).
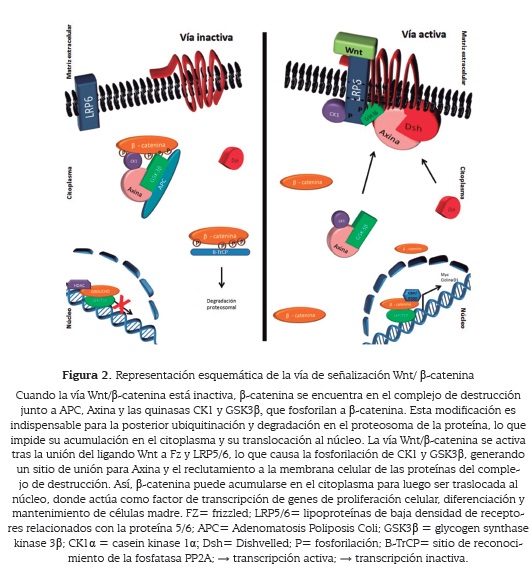
En el complejo de destrucción, la proteína APC se fosforila, generando un cambio conformacional que facilita su unión a β-catenina. La interacción APC-β-catenina permite la fosforilación de esta última por las quinasas del complejo. Adicionalmente, APC evita la defosforilación de β-catenina mediada por la fosfotasa 2A (PP2A), protegiendo el sitio de reconocimiento de β-TrCP (del inglés, Beta-transducing repeat containing protein), para mantener la unión de β-TrCP a la proteína β-catenina fosforilada y para su posterior degradación vía proteosomal (figura 2).
En el núcleo, APC facilita el reclutamiento del represor transcripcional CtBP al complejo β-catenina/LEF-1, regulando de forma negativa la transcripción de los genes blanco de la señalización Wnt (20). Teniendo en cuenta las propiedades de la proteína APC, el gen que la codifica es conocido como un gen supresor de tumores, puesto que funciona como un regulador negativo de la vía de señalización Wnt. A finales de la década de los 90 se describieron mutaciones en el gen APC en casos de cáncer colorrectal (21).
Cuando la vía está activada, la proteína β-catenina, luego de ser traslocada al núcleo, forma un complejo con miembros de la familia de proteínas de unión al ADN: TCF-1/LEF-1, 3, 4, lo que produce el desplazamiento de la proteína "Groucho", un co-represor transcripcional; β-catenina también facilita el reclutamiento de otras proteínas que favorecen la transcripción (co-activadores) como las acetilasas de histonas CBP/p300 (22) (figura 2). La formación del complejo β-catenina-TCF1/LEF1 y el reclutamiento de CBP/p300 regula positivamente la expresión de genes que codifican proteínas como Cyclin D1, c-Myc y metaloproteasas de matriz celular (MMP-2, 3, 7, 9, 13), entre otros (23).
β-CATENINA EN LA ADHESIóN CELULAR
La organización espacial y funcional de las células en los tejidos se determina por la adhesión célula-célula. Esta interacción es esencial para la morfogénesis y la polaridad apicobasal de las células y se establece por la formación del complejo cadherina-catenina (24).
Las cadherinas son glicoproteínas de 120 kDa que presentan tres grandes dominios: extracelular (N-terminal), transmembranal e intracelular (c-terminal), con funciones y propiedades diferentes. La región extracelular de las cadherinas presenta cinco dominios que median la adhesión celular dependiente de calcio, por interacción con dominios extracelulares de cadherinas de las células vecinas que permite la formación de un "cierre" o "cremallera" (molecular zipper).
La región C-terminal de la cadherina o región intracelular es la responsable de la unión de esta proteína al citoesqueleto mediante la interacción con β-catenina y a-catenina. Esta interacción permite estabilizar y reforzar la unión de los dominios extracelulares de cadherina, que es relativamente débil; esto se logra mediante la unión entre los dominios citoplásmicos de las proteína cadherina y actina, componente principal del citoesqueleto de la célula, lo que resulta en el agrupamiento lateral de los dominios extracelulares de la cadherina (25).
Ahora bien, las cadherinas no están unidas a la proteína actina en forma directa, sino a través de su interacción con las proteínas β-catenina y α-catenina. Se ha demostrado que α-catenina se asocia con los filamentos de actina para permitir establecer la posterior unión con cadherina (26).
En este complejo también participa la proteína EPLIN (por las siglas en inglés, epithelial protein lost in neoplasm). EPLIN es una proteína que potencia la unión y el agrupamiento de los filamentos de actina, clave en el interacción estable del complejo cadherina-catenina-Eplin y por tanto es un factor muy importante en la unión célula-célula (27, 28).
A este complejo macromolecular se une la proteína p120-catenina, miembro de la familia de proteínas con dominio "armadillo". p120-catenina juega un papel esencial en la estabilización del complejo de adhesión celular, gracias a que impide la internalización de la cadherina (29). Además p120-catenina cumple una función muy importante como factor de regulación de la GTPasa Rho, que participa en la dinámica del citoesqueleto. En un modelo in vitro se demostró la función de p120-catenina en la capacidad para aumentar el área de contacto para la unión célula-célula, mientras que β-catenina es un factor determinante en la adhesión celular fuerte (30). Por otro lado, se ha demostrado que la proteína p120-catenina interacciona con otro de los componentes del citoesqueleto, los microtúbulos. Esta interacción sería mediada por las proteínas PLEKHA7 (por las siglas en inglés, pleckstrin homology domain-containing) y Nezha (31, 32) (figura 3).
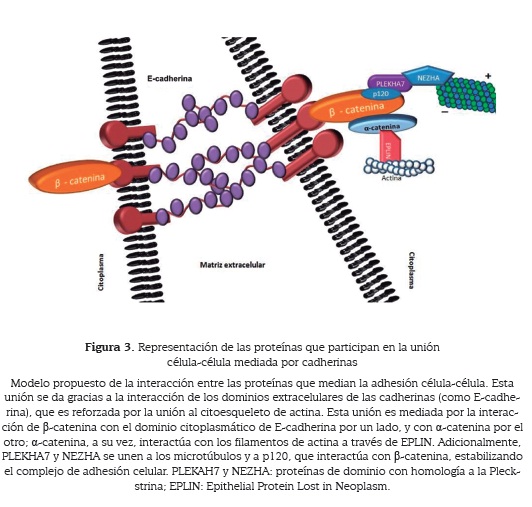
La fosforilación de p120-catenina y β-catenina es una modificación frecuente en el proceso de adhesión célula-célula mediada por cadherinas. Datos experimentales indican que la presencia de β-catenina en las uniones celulares está controlada por su fosforilación en el residuo Tyr-654; la modificación de este aminoácido en la última repetición "armadillo" de la proteína β-catenina disminuye la capacidad de unión a E-cadherina y por tanto afecta la formación de las uniones adherentes (4).
Además de su función como puente en el complejo cadherina/β-catenina/a-catenina, la proteína β-catenina es necesaria para facilitar el transporte de cadherinas del aparato de Golgi a la membrana plasmática (33). También es importante en la estabilización estructural de las uniones celulares y en la interacción proteína- proteína con otros factores como vinculina, componente de la matriz extracelular (34).
La modificación de este complejo para desmontar la uniones célula-célula hace parte de la fisiología celular; por ejemplo esta modificación es necesaria en la transición epitelial-mesenquimal, en procesos de diferenciación celular y de migración (35).
Sin embargo, alteraciones en el complejo de adhesión célula-célula por mutaciones en el gen CDH1, gen que codifica la proteína E-cadherina, han sido descritas para diferentes tipos de tumores, como carcinoma lobular de mama, cáncer gástrico de tipo difuso y carcinoma hepatocelular (36-38).
Las alteraciones del complejo también pueden ser consecuencia de cambios en la actividad del promotor. En el carcinoma hepatocelular se ha demostrado la hipermetilación del promotor del gen CDH1 y la subsecuente disminución en el nivel de expresión de la proteína E-cadherina (39).
Un estudio reciente permitió demostrar que en la carcinogénesis asociada a la infección crónica por el virus de la hepatitis C, la alteración epigenética del gen CDH1 estaba directamente relacionada con la replicación viral. En este contexto la infección por este virus ocasiona un aumento en la actividad de las ADN metil transferasas (DNMT), enzimas encargadas de la metilación del genoma celular. Una de las consecuencias de esta actividad es la metilación del promotor del gen CDH1 en las células de hepatoma Huh-7.5 y la ulterior interrupción en la transcripción, traducción y expresión de la proteína E-cadherina (40).
Cambios en el nivel de expresión de la proteína E-cadherina debida a mutaciones o a modificaciones epigenéticas del promotor del gen correspondiente entre otros, es uno de los eventos del proceso multi-etapas de la carcinogénesis relacionados con metástasis como resultado de la migración e invasión de las células tumorales a otros tejidos. Esta propiedad de las células tumorales está directamente relacionada con la pérdida del complejo de adhesión celular.
REGULACIóN DE LA VíA SEñALIZACIóN WNT/β- CATENINA EN CáNCER
La vía de señalización Wnt funciona en respuesta a la presencia de β-catenina, y es fundamental en el desarrollo embrionario y en la homeostasis tisular (23). Es altamente conservada a través de la evolución (41), lo que puede considerase una señal de su importancia. Mutaciones y cambios epigenéticos en la vía Wnt se han relacionado con defectos congénitos (42); además, esta vía está alterada en diversos tipos de cáncer, así como en enfermedades óseas y cardiovasculares (42, 43).
Estudios recientes evidencian mecanismos de alteración de los componentes de la vía de señalización Wnt/β-catenina, diferente a la regulación del gen CTNNB1; estas alteraciones generan hiperactivación de la vía Wnt, lo que puede resultar en un proceso de carcinogénesis.
La pérdida de expresión de algunos genes que codifican proteínas de la vía Wnt está relacionada con la estabilización de la proteína β-catenina, con la actividad continua de esta proteína como co-activador transcripcional y con la activación de la proliferación debido a la estimulación constante de la vía.
Es el caso del carcinoma colorrectal, en el cual se ha descrito que en el 85 % de los casos se presentan mutaciones en el gen APC; estas mutaciones afectan su función en el complejo de destrucción de β-catenina. Debido a esta alteración genética se observa acumulación citoplasmática de β-catenina en líneas celulares de cáncer colorectal, además de que la señalización Wnt está activada constitutivamente. Las mutaciones en el gen APC están generalmente dirigidas a alterar los dominios reguladores de β-catenina, aunque no necesariamente aquellos de unión a esta proteína (44).
Los sitios de unión a la proteína axina son críticos para la proteína APC y su rol en la vía de señalización, por lo cual la modificación de éstos altera los niveles de β-catenina y puede favorecer los procesos de carcinogénesis (45). La proteína axina fue originalmente identificada como un inhibidor de la señalización de Wnt en embriones de rana de la especie Xenopus. Diversos estudios in vitro e in vivo (Xenopus, Drosophila y células de mamíferos) aportaron evidencias del papel de la axina en la regulación de β-catenina (46).
Esta proteína también se conoce como proteína "andamio" del complejo de destrucción, gracias a que se une a las proteínas APC, β-catenina, GSK3β y Dishvelled (47). Se ha propuesto además que la proteína axina facilita la fosforilación de β-catenina y APC por acción de las quinasas GSK3β y CK1α (48). Es por esto que los genes Axin1 y Axin2 son considerados como genes supresores de tumores por la capacidad de la proteína axina de regular negativamente la señalización Wnt.
Mutaciones en los genes Axin1 y Axin2 constituyen un defecto genético en la vía de Wnt que contribuye al cáncer humano. También han sido descritos patrones de hipermetilación de secuencias promotoras en el gen APC y como se mencionó anteriormente en el gen CDH-1, lo que conlleva al silenciamiento de estos genes y a la ausencia de las proteínas correspondientes. Como consecuencia de la ausencia de estas proteínas, se altera la formación y eficiencia del complejo de destrucción y por ende a la estabilización y acumulación intracelular de β-catenina, favoreciendo así el desarrollo y progresión de tumores (49, 50).
Otros factores de vías de señalización diferentes a Wnt también pueden estar involucrados en la regulación de β-catenina mediante la fosforilación de esta proteína o por la regulación de otros componentes corriente arriba de la vía de señalización Wnt/β-catenina. En estudios in vitro se ha demostrado la interacción de β-catenina con miembros de la familia del receptor tirosina quinasa del factor de crecimiento epidermal (EGFR), como el receptor de crecimiento epidermal humano 2 (HER2); la fosforilación de β-catenina mediada por estos receptores modula la actividad transcripcional de β-catenina (51).
También pueden participar en la regulación de la vía Wnt las proteínas codificadas por los genes supresores de tumores p53 y PTEN (por las siglas en inglés, phosphatase and tensin homolog). Por ejemplo, la pérdida de expresión de PTEN resulta en la acumulación de β-catenina y en la activación transcripcional dependiente del TCF (52).
Y aunque no es claro el mecanismo molecular, la degradación de β-catenina puede ser regulada positivamente por la proteína p53; se sugiere un mecanismo indirecto, dado que no se ha podido demostrar la interacción proteína-proteína entre p53 y β-catenina. Esta regulación por p53 controla la acumulación de β-catenina, protegiendo a la célula de posibles eventos oncogénicos (53).
Recientemente se ha descrito la relación de la actividad de la telomerasa, subunidad enzimática que controla la longitud de los telómeros, y la proteína β-catenina; el promotor del gen TERT es directamente regulado por β-catenina, de tal manera que la acumulación de la proteína β-catenina, por mutaciones en el gen CTNNB1, pueden provocar un aumento en la expresión del gen TERT, una etapa importante en el proceso de carcinogénesis (54).
ALTERACIÓN GENÉTICA DEL GEN CTNNB1 EN CÁNCER
En neoplasias caracterizadas por alteración de la vía de señalización Wnt/ β-catenina son frecuentes las mutaciones en el gen CTNNB1. Los cambios en la secuencia de este gen más reportados se localizan en el exón 3, que codifica parte del dominio N-terminal de la proteína β-catenina en el cual se ubican los residuos de serina y treonina, Ser45, Thr41, Ser37 y Ser33, blanco de las quinasas GSK3β y CK1α (55). Estas mutaciones producen cambios conformacionales en la proteína que impiden su fosforilación, lo cual resulta en la estabilización de β-catenina y en su acumulación citoplasmática, posterior traslocación al núcleo y en consecuencia, transcripción de los genes blanco de la vía Wnt/β-catenina (20, 23). Estas mutaciones en el exón 3 se han descrito en diferentes tipos de cáncer incluyendo cáncer de colon, de hígado, de ovario, en melanoma y en tumores desmoides (figura 1A) (20, 23).
El hepatoblastoma, cáncer primario de hígado, que se presenta principalmente en niños, es una neoplasia que se caracteriza por una alta frecuencia de mutaciones en el gen CTNNB1; es así como el 75 al 80 % de los casos de este tumor presentan mutaciones en este gen y así como en el modelo murino de hepatoblastoma. En otro cáncer primario de hígado, el carcinoma hepatocelular, se han descrito mutaciones en el exón 3 del gen CTNNB1 hasta en un 44 % en los casos (56, 57). En un estudio reciente realizado con muestras de tumores primarios de origen desconocido, se demostró que el 20.7% de los tumores presentaban mutaciones en los exones 3, 4 y 5 del gen CTNNB1. Y en el 19.5% de los casos estas mutaciones estaban relacionadas con la activación de la vía y además se demostró una asociación significativa con mal pronóstico de los pacientes (58).
Las mutaciones S45A, T41A, S37A, S33A aumentan el nivel de capacidad transformante de las células; adicionalmente también pueden tener efecto tanto en la proliferación como en la migración celular. En ensayos in vitro con células MDCK, una línea celular no transformada de células epiteliales con expresión de cadherina y componentes de la vía de señalización Wnt "wild type", se observó que las mutaciones S37A y S33A generan disminución en la densidad celular, mientras que las mutaciones T41A y S45A aumentan el número de células en la fase G0 del ciclo celular. Adicionalmente, en este modelo se demostró que la mutación S45A aumenta la migración celular y los niveles de ARNm de ciclina D1, un gen blanco de la activación transcripcional por β-catenina (59).
REGULACIÓN DE LA ADHESIÓN CELULAR MEDIADA POR CADHERINAS EN CÁNCER
Las alteraciones del complejo cadherina-catenina se han relacionado con una pérdida de la adhesión celular, lo que lleva al desarrollo de célula con gran capacidad invasiva (60, 61).
La estabilización de β-catenina en células tumorales juega un papel muy importante tanto en el desarrollo como en la progresión del tumor, con incremento de la habilidad metastásica de éstas células; además, ha sido asociada con una mayor capacidad invasiva de las células tumorales afectando el equilibrio celular, promoviendo la proliferación y diferenciación tumoral en cáncer de colon, estómago, esófago, hígado, cuello, pulmón y seno. Tanto la acumulación de β-catenina como su localización nuclear están relacionadas con un peor pronóstico del cáncer colorrectal (5).
Se ha demostrado in vitro la capacidad de β-catenina en mantener el fenotipo maligno en líneas celulares de hepatoma y carcinoma hepatocelular (HuH-7 y HepG2). Estas líneas celulares se caracterizan por la expresión de β-catenina mutada. La regulación negativa de la expresión de la proteína por siRNA- β-catenina, produce una disminución en la expresión de genes blanco como c-myc y Ciclina D1, además de una disminución en la proliferación celular (62).
Estudios recientes en diferentes tipos tumorales indican que las alteraciones en el gen CTNNB1 puede ser un evento temprano durante el desarrollo del cáncer de colon, gástrico, urogenital y CHC. Además, se ha evidenciado el papel de estas mutaciones en la proliferación celular y en la inducción de apoptosis en células hepáticas transfectadas con el gen CTNNB1 mutado y en ratones transgénicos para la proteína β-catenina mutada (63, 64).
La alteración de la adhesión celular mediada por E-cadherina se considera un paso clave en la progresión hacia la fase de un tumor invasivo (65). Los mecanismos responsables de estos cambios en la adhesión incluyen mutaciones en el gen CDH1, que alteran la capacidad de adhesión de la proteína (66), la hipermetilación del promotor de este gen (67) o una combinación de mutaciones en un alelo y silenciamiento génico por metilación del promotor del alelo restante (37). Adicionalmente se ha descrito la pérdida de la expresión de E-cadherina debido a su represión transcripcional (68-70).
Varios factores de transcripción están implicados en la represión del gen CDH1 como las proteínas de la familia "dedos de zinc" Slug / Snail, F1/ZEB1, SIP-1, y el factor base "hélice-bucle-hélice" E12/E47 (69, 71, 72). La represión transcripcional del gen CDH1 y los subsecuentes cambios morfológicos en las células ocurren durante la transición epitelio mesenquimal en el desarrollo embrionario, cuando las células epiteliales se diferencian y durante la migración de células de la cresta neural del neuroectodermo (73).
miRNAs
Se ha reportado también la identificación de la regulación de la vía Wnt/β-catenina por microARNs (miRNAs, por sus siglas en inglés); los miRNAs son cadenas de ácido ribonucleico de 18 a 24 nucleótidos de largo, no-codificantes, que participan en el silenciamiento del ARNm y la regulación post-transcripcional de la expresión génica. Este mecanismo de silenciamiento es un potente regulador de la expresión génica y de procesos como la inflamación y la oncogénesis. El análisis de la expresión de miRNA en diferentes neoplasias ha permitido definir el perfil de miRNA característico de cada tumor y su importancia en el desarrollo, así como la relación de algunos factores de riesgo, miRNA y la neoplasia (74)
Una evidencia de la interacción de factor de riesgo y miRNA fue aportada por un estudio reciente en muestras de tejido hepático provenientes de pacientes con infección crónica por VHC y pacientes con carcinoma hepatocelular asociado a la infección por VHC. El análisis de estas muestras permitió demostrar un aumento en el nivel de expresión de miRNA-155, comparado con lo observado en tejidos provenientes de pacientes con esteato hepatitis no alcohólica (NASH, por su sigla en inglés). Este hallazgo fue corroborado por los resultados observados in vitro en una línea de hepatoma humano infectada con VHC, segunda evidencia de que miRNA-155 es regulado por la infección por VHC. Este miRNA promueve la proliferación de hepatocitos e inhice la apoptosis gracias a la activación de la vía Wnt/β-catenina, activación que depende del silenciamiento del gen APC.
Como se describió anteriormente, la proteína APC hace parte del complejo de destrucción, con las proteínas GSK3β, CK1α y axina, clave en la regulación de β-catenina y por tanto de esta vía. Adicionalmente, los autores demuestran la relación entre la respuesta inflamatoria y la oncogénesis en este modelo, dado que el miRNA-155 es regulado positivamente por la respuesta inflamatoria consecuencia de la infección por VHC y esto conlleva a la activación de la vía Wnt lo que promueve la hepato-oncogénesis (75).
De otra parte se ha demostrado la regulación negativa del miRNA-432 en el carcinoma hepatocelular, lo que está asociado con la proliferación y tumorigenicidad de los hepatocitos. Estos hallazgos fueron descritos en tejido hepático proveniente de pacientes con diagnóstico de carcinoma hepatocelular, comparado con el nivel de mi-RNA en tejido hepático no tumoral proveniente de los mismos pacientes. Los autores demuestran además en un modelo in vitro que este miRNA-432 silencia los genes que codifican las proteínas LRP6, TRIM29 y Pygo2 que hacen parte de esta vía de señalización; y como consecuencia, se inhibe la proliferación celular.
La sobre expresión de estas tres proteínas ha sido descrita en diferentes tipos de tumor. De tal manera, miRNA-432 representa un miRNA supresor de tumores y un importante blanco terapéutico para el manejo de neoplasias como el carcinoma hepatocelular (76). En cáncer ovárico también se ha demostrado una regulación negativa de este miRNA (77) . El miR-601 también se podría considerar un miRNA supresor de tumores teniendo en cuenta que la inhibición de este miR-610 activa la vía Wnt/β-catenina, mediante la supresión de LRP6 y TBL1X y promueve la proliferación celular y tumorigenicidad in vitro como in vivo (78). En tejidos de carcinoma hepatocelular se ha demostrado la regulación negativa de miRNA-610 y además se ha relacionado con la progresión de la enfermedad y mal pronóstico (78).
En células de cáncer colorrectal se ha demostrado que el miRNA-29c regula negativamente la vía Wnt/β-catenina a través de las proteínas GNA13 y PTP4A y esto genera una inhibición significativa de la capacidad de migración e invasión de las células in vitro y de metástasis in vivo. También se demostró que miRNA-29c inhibe la transición epitelial-mesenquimal de las células (79).
Al igual que la vía de señalización Wnt/β- catenina, el complejo Cadherina- β-catenina es regulado por miRNAs. El blanco del miRNA-9 es el gen que codifica la proteína E-cadherina; el silenciamiento de este gen y por tanto de la proteína E-cadherina repercute en la estabilidad de la proteína β-catenina en citoplasma y posterior traslocación a núcleo. En muestras de carcinoma de células escamosas se ha demostrado un aumento en la expresión de miRNA-9, que se asocia con la migración celular debido a la regulación negativa de E-cadherina (80)
CONCLUSIONES
La vía Wnt/ β-catenina, que funciona en respuesta al coactivador transcripcional β-catenina, tiene un papel fundamental en la regulación de la expresión de proteínas claves en el desarrollo embrionario, organogénesis y homeostasis. Además de participar en esta importante vía, la proteína β-catenina también participa en la organización, morfogénesis y polaridad apicobasal de las células, gracias a que hace parte del complejo de adhesión célula-célula cadherina- β-catenina- α-catenina.
La vía Wnt/β-catenina es altamente conservada a nivel evolutivo, lo que demuestra su importancia a nivel celular. La hiperactivación de esta vía se ha asociado con la carcinogénesis, debido a que la estabilización o no degradación de β-catenina hace que actúe continuamente como regulador de la transcripción y como activador de la proliferación celular. Diversos estudios han presentado evidencias de desregulación de la vía Wnt/ β-catenina en carcinogénesis y las implicaciones de la alteración de otras de las proteínas que hacen parte de esta importante vía de señalización celular, como APC y axina. Uno de los mecanismos más comunes de alteración de esta vía es la acumulación de mutaciones en el gen CTNNB1, que codifica para β-catenina, que generan cambios en la secuencia de aminoácidos. Estas mutaciones impiden la fosforilación de la proteína y por tanto el control de la acumulación de ésta en el citoplasma y su posterior translocación al núcleo. Por otro lado, algunas mutaciones en el gen de β-catenina causan un aumento en la capacidad transformarte de las células, así como en la proliferación y la migración celular.
Mutaciones que afecten a otras proteínas de la vía, especialmente mutaciones de pérdida de función en APC o axina, también contribuyen a la hiperactividad de la vía al impedir el correcto funcionamiento del complejo de degradación. Finalmente, se ha encontrado que proteínas como p53 y PTEN pueden regular negativamente a β-catenina, a pesar de no ser parte de la vía Wnt/ β-catenina.
Con respecto a la adhesión celular, la estabilización de β-catenina, que media la interacción entre cadherinas y actina, se ha asociado con mayor capacidad invasiva, proliferación y diferenciación celular en varios tipos de tumores.
Finalmente, se han realizado varios estudios sobre la desregulación de miRNAs en cáncer y su relación con la vía de sealización Wnt/β-catenina o la adhesión celular mediada por esta proteína.
La exploración de la vía y de cada uno de los procesos implicados en su desregulación permitiría descifrar la clave del balance entre las funciones β-catenina en la adhesión celular y como factor de transcripción, y de esta manera definir blancos terapéuticos para el control de neoplasias como cáncer colorrectal y carcinoma hepatocelular, en las cuales la alteración de Wnt/β-catenina es un evento trascendental en el proceso tumoral.
AGRADECIMIENTOS
Esta publicación hace parte del proyecto 111540820471 financiado por el Departamento Nacional de Ciencia Innovación y Tecnología, Colciencias, y del proyecto de Sostenibilidad de la Vicerrectoría de Investigación de la Universidad de Antioquia.
REFERENCIAS
1. Fu Y, Zheng S, An N, Athanasopoulos T, Popplewell L, Liang A, et al. Beta-catenin as a potential key target for tumor suppression. Int J Cancer. 2011;129(7):1541-51. [ Links ]
2. Katanaev VL. The Wnt/Frizzled GPCR signaling pathway. Biochemistry (Mosc). 2010;75(12):1428-34. [ Links ]
3. Camilli TC, Weeraratna AT. Striking the target in Wnt-y conditions: intervening in Wnt signaling during cancer progression. Biochem Pharmacol. 2010;80(5):702-11. [ Links ]
4. Piedra J, Martinez D, Castano J, Miravet S, Dunach M, de Herreros AG. Regulation of beta-catenin structure and activity by tyrosine phosphorylation. J Biol Chem. 2001;276(23):20436-43. [ Links ]
5. Akiyama T. Wnt/beta-catenin signaling. Cytokine Growth Factor Rev. 2000;11(4):273-82. [ Links ]
6. Cowin P, Rowlands TM, Hatsell SJ. Cadherins and catenins in breast cancer. Curr Opin Cell Biol. 2005;17(5):499-508. [ Links ]
7. Huber AH, Nelson WJ, Weis WI. Three-dimensional structure of the armadillo repeat region of beta-catenin. Cell. 1997;90(5):871-82. [ Links ]
8. MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17(1):9-26. [ Links ]
9. Angers S, Moon RT. Proximal events in Wnt signal transduction. Nat Rev Mol Cell Biol. 2009;10(7):468-77. [ Links ]
10. Gumbiner BM. Signal transduction of betacatenin. Curr Opin Cell Biol. 1995;7(5):634- 40. [ Links ]
11. Nusse R, Varmus HE. Wnt genes. Cell. 1992;69(7):1073-87. [ Links ]
12. Korswagen HC, Herman MA, Clevers HC. Distinct beta-catenins mediate adhesion and signalling functions in C. elegans. Nature. 2000;406(6795):527-32. [ Links ]
13. Mao J, Wang J, Liu B, Pan W, Farr GH, 3rd, Flynn C, et al. Low-density lipoprotein receptor-related protein-5 binds to Axin and regulates the canonical Wnt signaling pathway. Mol Cell. 2001;7(4):801-9. [ Links ]
14. He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281(5382):1509-12. [ Links ]
15. Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R, et al. The cyclin D1 gene is a target of the beta-catenin/ LEF-1 pathway. Proc Natl Acad Sci U S A. 1999;96(10):5522-7. [ Links ]
16. Roose J, Huls G, van Beest M, Moerer P, van der Horn K, Goldschmeding R, et al. Synergy between tumor suppressor APC and the beta-catenin-Tcf4 target Tcf1. Science. 1999;285(5435):1923-6. [ Links ]
17. Takahashi Y, Nishikawa M, Takakura Y. Suppression of tumor growth by intratumoral injection of short hairpin RNA-expressing plasmid DNA targeting beta-catenin or hypoxia- inducible factor 1alpha. J Control Release. 2006;116(1):90-5. [ Links ]
18. Luo W, Peterson A, Garcia BA, Coombs G, Kofahl B, Heinrich R, et al. Protein phosphatase 1 regulates assembly and function of the beta-catenin degradation complex. EMBO J. 2007;26(6):1511-21. [ Links ]
19. Taelman VF, Dobrowolski R, Plouhinec JL, Fuentealba LC, Vorwald PP, Gumper I, et al. Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell. 2010;143(7):1136-48. [ Links ]
20. Fodde R, Brabletz T. Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr Opin Cell Biol. 2007;19(2):150-8. [ Links ]
21. Bienz M. APC: the plot thickens. Curr Opin Genet Dev. 1999;9(5):595-603. [ Links ]
22. Tillhon M, Cazzalini O, Nardo T, Necchi D, Sommatis S, Stivala LA, et al. p300/CBP acetyl transferases interact with and acetylate the nucleotide excision repair factor XPG. DNA Repair (Amst). 2012. [ Links ]
23. Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781-810. [ Links ]
24. Yamada S, Pokutta S, Drees F, Weis WI, Nelson WJ. Deconstructing the cadherin- catenin-actin complex. Cell. 2005;123(5):889-901. [ Links ]
25. Jamora C, Fuchs E. Intercellular adhesion, signalling and the cytoskeleton. Nat Cell Biol. 2002;4(4):E101-8. [ Links ]
26. Rimm DL, Koslov ER, Kebriaei P, Cianci CD, Morrow JS. Alpha 1(E)-catenin is an actinbinding and -bundling protein mediating the attachment of F-actin to the membrane adhesion complex. Proc Natl Acad Sci U S A. 1995;92(19):8813-7. [ Links ]
27. Abe K, Takeichi M. EPLIN mediates linkage of the cadherin catenin complex to F-actin and stabilizes the circumferential actin belt. Proc Natl Acad Sci U S A. 2008;105(1):13-9. [ Links ]
28. Meng W, Takeichi M. Adherens junction: molecular architecture and regulation. Cold Spring Harb Perspect Biol. 2009;1(6):a002899. [ Links ]
29. Chiasson CM, Wittich KB, Vincent PA, Faundez V, Kowalczyk AP. p120-catenin inhibits VE-cadherin internalization through a Rhoindependent mechanism. Mol Biol Cell. 2009;20(7):1970-80. [ Links ]
30. Oas RG, Nanes BA, Esimai CC, Vincent PA, García AJ, Kowalczyk AP. p120-catenin and ß-catenin differentially regulate cadherin adhesive function. Mol Biol Cell. 2013;24(6):704-14. [ Links ]
31. Meng W, Mushika Y, Ichii T, Takeichi M. Anchorage of microtubule minus ends to adherens junctions regulates epithelial cell-cell contacts. Cell. 2008;135(5):948-59. [ Links ]
32. Ishiyama N, Lee SH, Liu S, Li GY, Smith MJ, Reichardt LF, et al. Dynamic and static interactions between p120 catenin and E-cadherin regulate the stability of cell-cell adhesion. Cell. 2010;141(1):117-28. [ Links ]
33. Miranda KC, Khromykh T, Christy P, Le TL, Gottardi CJ, Yap AS, et al. A dileucine motif targets E-cadherin to the basolateral cell surface in Madin-Darby canine kidney and LLC-PK1 epithelial cells. J Biol Chem. 2001;276(25):22565-72. [ Links ]
34. Peng X, Cuff LE, Lawton CD, DeMali KA. Vinculin regulates cell-surface E-cadherin expression by binding to beta-catenin. J Cell Sci. 2010;123(Pt 4):567-77. [ Links ]
35. Conacci-Sorrell M, Simcha I, Ben-Yedidia T, Blechman J, Savagner P, Ben-Ze'ev A. Autoregulation of E-cadherin expression by cadherin-cadherin interactions: the roles of beta-catenin signaling, Slug, and MAPK. J Cell Biol. 2003;163(4):847-57. [ Links ]
36. Berx G, Becker KF, Hofler H, van Roy F. Mutations of the human E-cadherin (CDH1) gene. Hum Mutat. 1998;12(4):226-37. [ Links ]
37. Machado JC, Oliveira C, Carvalho R, Soares P, Berx G, Caldas C, et al. E-cadherin gene (CDH1) promoter methylation as the second hit in sporadic diffuse gastric carcinoma. Oncogene. 2001;20(12):1525-8. [ Links ]
38. Tsuda H, Zhang WD, Shimosato Y, Yokota J, Terada M, Sugimura T, et al. Allele loss on chromosome 16 associated with progression of human hepatocellular carcinoma. Proc Natl Acad Sci U S A. 1990;87(17):6791-4. [ Links ]
39. Kanai Y, Ushijima S, Hui AM, Ochiai A, Tsuda H, Sakamoto M, et al. The E-cadherin gene is silenced by CpG methylation in human hepatocellular carcinomas. Int J Cancer. 1997;71(3):355-9. [ Links ]
40. Park J, Jang KL. Hepatitis C virus represses Ecadherin expression via DNA methylation to induce epithelial to mesenchymal transition in human hepatocytes. Biochem Biophys Res Commun. 2014;446(2):561-7. [ Links ]
41. Moon R. Wnt/β-Catenin Pathway. Sci. STKE; 2008. p. cm1. [ Links ]
42. Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127(3):469-80. [ Links ]
43. MacDonald BT, Yokota C, Tamai K, Zeng X, He X. Wnt signal amplification via activity, cooperativity, and regulation of multiple intracellular PPPSP motifs in the Wnt co-receptor LRP6. J Biol Chem. 2008;283(23):16115-23. [ Links ]
44. Polakis P. The oncogenic activation of beta-catenin. Curr Opin Genet Dev. 1999;9(1):15-21. [ Links ]
45. Kawahara K, Morishita T, Nakamura T, Hamada F, Toyoshima K, Akiyama T. Downregulation of beta-catenin by the colorectal tumor suppressor APC requires association with Axin and beta-catenin. J Biol Chem. 2000;275(12):8369-74. [ Links ]
46. Farr GH, 3rd, Ferkey DM, Yost C, Pierce SB, Weaver C, Kimelman D. Interaction among GSK-3, GBP, axin, and APC in Xenopus axis specification. J Cell Biol. 2000;148(4):691-702. [ Links ]
47. Peifer M, Polakis P. Wnt signaling in oncogenesis and embryogenesis--a look outside the nucleus. Science. 2000;287(5458):1606-9. [ Links ]
48. Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S, Kikuchi A. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J. 1998;17(5):1371-84. [ Links ]
49. Kwon GY, Yoo BC, Koh KC, Cho JW, Park WS, Park CK. Promoter methylation of E-cadherin in hepatocellular carcinomas and dysplastic nodules. J Korean Med Sci. 2005;20(2):242-7. [ Links ]
50. Katoh H, Shibata T, Kokubu A, Ojima H, Fukayama M, Kanai Y, et al. Epigenetic instability and chromosomal instability in hepatocellular carcinoma. Am J Pathol. 2006;168(4):1375-84. [ Links ]
51. Behari J. The Wnt/beta-catenin signaling pathway in liver biology and disease. Expert Rev Gastroenterol Hepatol. 2010;4(6):745- 56. [ Links ]
52. Persad S, Troussard AA, McPhee TR, Mulholland DJ, Dedhar S. Tumor suppressor PTEN inhibits nuclear accumulation of beta-catenin and T cell/lymphoid enhancer factor 1-mediated transcriptional activation. J Cell Biol. 2001;153(6):1161-74. [ Links ]
53. Mei Y, Xie C, Xie W, Wu Z, Wu M. Siah-1S, a novel splice variant of Siah-1 (seven in absentia homolog), counteracts Siah-1-mediated downregulation of beta-catenin. Oncogene. 2007;26(43):6319-31. [ Links ]
54. Hoffmeyer K, Raggioli A, Rudloff S, Anton R, Hierholzer A, Del Valle I, et al. Wnt/ beta-catenin signaling regulates telomerase in stem cells and cancer cells. Science. 2012;336(6088):1549-54. [ Links ]
55. Lee HS, Park MH, Yang SJ, Park KC, Kim NS, Kim YS, et al. Novel candidate targets of Wnt/beta-catenin signaling in hepatoma cells. Life Sci. 2007;80(7):690-8. [ Links ]
56. Hsu HC, Jeng YM, Mao TL, Chu JS, Lai PL, Peng SY. Beta-catenin mutations are associated with a subset of low-stage hepatocellular carcinoma negative for hepatitis B virus and with favorable prognosis. Am J Pathol. 2000;157(3):763-70. [ Links ]
57. Laurent-Puig P, Legoix P, Bluteau O, Belghiti J, Franco D, Binot F, et al. Genetic alterations associated with hepatocellular carcinomas define distinct pathways of hepatocarcinogenesis. Gastroenterology. 2001;120(7):1763-73. [ Links ]
58. Pentheroudakis G, Kotteas EA, Kotoula V, Papadopoulou K, Charalambous E, Cervantes A, et al. Mutational profiling of the RAS, PI3K, MET and b-catenin pathways in cancer of unknown primary: a retrospective study ofthe Hellenic Cooperative Oncology Group. Clin Exp Metastasis. 2014;31(7):761-9. [ Links ]
59. Provost E, Yamamoto Y, Lizardi I, Stern J, D'Aquila TG, Gaynor RB, et al. Functional correlates of mutations in beta-catenin exon 3 phosphorylation sites. J Biol Chem. 2003;278(34):31781-9. [ Links ]
60. Stepniak E, Radice GL, Vasioukhin V. Adhesive and signaling functions of cadherins and catenins in vertebrate development. Cold Spring Harb Perspect Biol. 2009;1(5):a002949. [ Links ]
61. Yuan S, Shi Y, Tang SJ. Wnt Signaling in the Pathogenesis of Multiple Sclerosis-Associated Chronic Pain. J Neuroimmune Pharmacol. 2012. [ Links ]
62. Sangkhathat S, Kusafuka T, Miao J, Yoneda A, Nara K, Yamamoto S, et al. In vitro RNA interference against beta-catenin inhibits the proliferation of pediatric hepatic tumors. Int J Oncol. 2006;28(3):715-22. [ Links ]
63. Shang XZ, Zhu H, Lin K, Tu Z, Chen J, Nelson DR, et al. Stabilized beta-catenin promotes hepatocyte proliferation and inhibits TNFalpha-induced apoptosis. Lab Invest. 2004;84(3):332-41. [ Links ]
64. Vanden Heuvel GB, Brantley JG, Alcalay NI, Sharma M, Kemeny G, Warolin J, et al. Hepatomegaly in transgenic mice expressing the homeobox gene Cux-1. Mol Carcinog. 2005;43(1):18-30. [ Links ]
65. Christofori G, Semb H. The role of the celladhesion molecule E-cadherin as a tumoursuppressor gene. Trends Biochem Sci. 1999;24(2):73-6. [ Links ]
66. Hajra KM, Fearon ER. Cadherin and catenin alterations in human cancer. Genes Chromosomes Cancer. 2002;34(3):255-68. [ Links ]
67. Hennig G, Behrens J, Truss M, Frisch S, Reichmann E, Birchmeier W. Progression of carcinoma cells is associated with alterations in chromatin structure and factor binding at the E-cadherin promoter in vivo. Oncogene. 1995;11(3):475-84. [ Links ]
68. Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2(2):76-83. [ Links ]
69. Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, et al. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell. 2001;7(6):1267-78. [ Links ]
70. Hajra KM, Chen DY, Fearon ER. The SLUG zincfinger protein represses E-cadherin in breast cancer. Cancer Res. 2002;62(6):1613-8. [ Links ]
71. Nieto MA. The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol. 2002;3(3):155-66. [ Links ]
72. Bolos V, Peinado H, Perez-Moreno MA, Fraga MF, Esteller M, Cano A. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors. J Cell Sci. 2003;116(Pt 3):499-511. [ Links ]
73. Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2(6):442-54. [ Links ]
74. Szabo G, Bala S. MicroRNAs in liver disease. Nat Rev Gastroenterol Hepatol. 2013;10(9):542-52. [ Links ]
75. Zhang Y, Wei W, Cheng N, Wang K, Li B, Jiang X, et al. Hepatitis C virus-induced up-regulation of microRNA-155 promotes hepatocarcinogenesis by activating Wnt signaling. Hepatology. 2012;56(5):1631-40. [ Links ]
76. Jiang N, Chen W-J, Zhang J-W, Xu C, Zeng X-C, Zhang T, et al. Downregulation of miR- 432 activates Wnt/β-catenin signaling and promotes human hepatocellular carcinoma proliferation. Oncotarget; 2015. p. 7866-79. [ Links ]
77. Kim YW, Kim EY, Jeon D, Liu JL, Kim HS, Choi JW, et al. Differential microRNA expression signatures and cell type-specific association with Taxol resistance in ovarian cancer cells. Drug Des Devel Ther. 2014;8:293-314. [ Links ]
78. Zeng X, Liu F, Yan R, Yi H, Zhang T, Wang G, et al. Downregulation of miR-610 promotes proliferation and tumorigenicity and activates Wnt/β-catenin signaling inhuman hepatocellular carcinoma. Mol Cancer 2014. [ Links ]
79. Zhang JX, Mai SJ, Huang XX, Wang FW, Liao YJ, Lin MC, et al. MiR-29c mediates epithelial- to-mesenchymal transition in human colorectal carcinoma metastasis via PTP4A and GNA13 regulation of β-catenin signaling. Ann Oncol. 2014;25(11):2196- 204. [ Links ]
80. Song Y, Li J, Zhu Y, Dai Y, Zeng T, Liu L, et al. MicroRNA-9 promotes tumor metastasis via repressing E-cadherin in esophageal squamous cell carcinoma. Oncotarget. 2014;5(22):11669-80. [ Links ]
Recibido: junio 19 de 2012. Revisado: octubre 11 de 2012. Aceptado: octubre 16 de 2012
Forma de citar: Vivas CA, Ríos JJ, Romero HA. Pólipos endometriales, fisiopatología y factores de riesgo. Rev CES Med 2012; 26(2): 175-184
