










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkCES Medicina
versión impresa ISSN 0120-8705
CES Med. vol.29 no.2 Medellín jul./dic. 2015
Linfocitosis monoclonal de células B: una revisión de aspectos generales
Monoclonal B-cell lymphocytosis: a review of general points
ROSSANA VILLEGAS-GRACIA1, CATALINA FRANCO-ALZATE 2, PATRICIA JARAMILLO-ARBELÁEZ 3
1 Bacterióloga. Especialista en Hematología en el laboratorio clínico y manejo del banco de sangre. MSc Microbiología y Bioanálisis énfasis hematología. Docente programa de Bacteriología, Universidad de Córdoba. Grupo de investigaciones Microbiológicas y Biomédicas de Córdoba (GIMBIC). Medellín, Colombia. rossanvillegas7@hotmail.com.
2 Médica y cirujana. Especialista en Patología. Docente Facultad de Medicina. Universidad CES. Patóloga laboratorio PROLAB SAS. Medellín, Colombia.
3 Bacterióloga y Laboratorista Clínica. Especialista en Hematología. MSc Microbiología y Bioanálisis énfasis hematología. Docente Escuela de Microbiología, Universidad de Antioquia. Grupo de Hematopatología Molecular (HEMO). Medellín, Colombia.
Forma de citar: Villegas-Gracia R, Franco-Alzate C, Jaramillo-Arbeláez P. Linfocitosis monoclonal de células B: una revisión de aspectos generales. Rev CES Med 2015;29(2): 227-238
Recibido en: septiembre 17 de 2014. Revisado en: mayo 30 de 2015. Aceptado en: octubre 2 de 2015
RESUMEN
Introducción: la linfocitosis monoclonal de células B es una condición que se caracteriza por tener menos de 5x109/L células B clonales en la sangre periférica, en ausencia de signos clínicos o síntomas de un trastorno linfoproliferativo crónico de células B.
Métodos: se realizó una búsqueda de artículos publicados en bases de datos multidisciplinarias y específicas de las áreas de la salud como Pubmed, Ovid, Science Direct, SciELO, Scopus y Embase; se emplearon términos relacionados con el tema como Monoclonal B lymphocytosis, Monoclonal B cell lymphocytosis and chronic lymphocytic leukemia, Monoclonal B-cell lymphocytosis, chronic lymphocytic leukemia, con su contraparte en español; además se incluyeron algunos capítulos de libros. Se seleccionaron los artículos que cumplían con criterios de calidad metodológica y de contenido.
Resultados: diferentes estudios basados en grandes series hospitalarias de linfocitosis monoclonal de células B, han puesto de manifiesto un riesgo más elevado de los sujetos con la entidad de progresar a leucemia linfocítica crónica. Algunos investigadores afirman que la mayoría de las personas con linfocitosis monoclonal de células B tipo leucemia linfocítica crónica no desarrollarán enfermedad progresiva en un período de cinco años aproximadamente, el 75 % estará vivo con un recuento de linfocitos estable y entre el 1 y 4 % por año desarrollará progresivamente una enfermedad que requiere tratamiento.
Conclusiones: existe gran variedad en la prevalencia de linfocitosis monoclonal de células B, dependiente en gran medida de las características de la población examinada y de los métodos de detección utilizados para su identificación. Los factores de riesgo que determinan el paso de linfocitosis monoclonal de células B a leucemia linfocítica crónica están aún por determinarse; sin embargo, diversos estudios han reportado que la edad, el sexo, los antecedentes familiares de leucemia linfocítica crónica, la presencia de algunos polimorfismos de nucleótido simple y marcadores como ZAP 70, CD38 y CD49d, podrían estar relacionados con el riesgo de desarrollar la enfermedad. Sin embargo, hasta ahora el factor mejor definido de progresión es el recuento absoluto de células B.
PALABRAS CLAVE: Linfocitosis, Monoclonal, Células B, Leucemia, linfocítica crónica.
ABSTRACT
Introduction: monoclonal lymphocytosis of B cells is a condition characterized as having less than 5x109/L clonal B cells in the peripheral blood, in the absence of clinical signs or symptoms of a chronic B cell lymphoproliferative disorder
Methods: a search of articles published in multidisciplinary databases and specific areas of health, such as PubMed , Ovid , Science Direct, SciELO, Scopus and Embase data was performed; also some book chapters were included. Terms related with the topic were used like Monoclonal B lymphocytosis, Monoclonal B cell lymphocytosis, and chronic lymphocytic leukemia, Monoclonal B-cell lymphocytosis, chronic lymphocytic leukemia. Articles that met criteria for methodological quality and content were selected.
Results: different studies based on large hospital series of MBL, have manifested a higher risk of subjects with MBL progressing to B-CLL. Some researchers claim that most people with MBL type LLC will not develop progressive disease over a period of about 5 years, 75 % will be alive with a stable count of lymphocytes and between 1 % and 4 % per year will gradually develop a disease requiring treatment.
Conclusions: there is great variation in the prevalence of MBL, dependent largely on the characteristics of the study population and the detection methods used for identification. Methods using increasingly sensitive detection has led to the identification of increasingly smaller MBL clones. Risk factors that determine the passage of monoclonal B-cell lymphocytosis to chronic lymphocytic leukemia are yet to be determined, however, several studies have reported that the age, sex, family history of CLL, the presence of some single nucleotide polymorphisms and markers such as ZAP 70, CD38 and CD49d, could be related to the risk of developing the disease. But so far the best defined progression factor is the absolute B cell count.
KEY WORDS: Lymphocytosis, Monoclonal, B cells, Chronic lymphocytic leukemia.
INTRODUCCIÓN
El presente artículo tiene como objetivo mostrar los aspectos más relevantes de la linfocitosis monoclonal de células B. Para este fin se realizó una búsqueda de artículos publicados en bases de datos multidisciplinarias y específicas de las áreas de la salud, como Pubmed, Ovid, Science Direct, SciELO, Scopus y Embase y se emplearon términos relacionados con el tema como:
Monoclonal B lymphocytosis, Monoclonal B cell lymphocytosis and chronic lymphocytic leukemia, Monoclonal B-cell lymphocytosis, chronic lymphocytic leukemia, con su contraparte en español; se incluyeron algunos capítulos de libros. Fueron seleccionados los artículos que cumplían con criterios de calidad metodológica y de contenido.
La linfocitosis absoluta se define como la existencia de más de 4x109/L linfocitos en la sangre periférica. Esta alteración puede encontrarse en forma aguda o crónica. Algunos casos de linfocitosis aguda se observan en enfermedades infecciosas, sobre todo de tipo viral, como la infección por citomegalovirus, por virus de Epstein Bar y adenovirus (1).
Los estados que se asocian con linfocitosis crónica son principalmente de tipo primario y dentro de estos se encuentran las neoplasias linfoproliferativas crónicas, entre las que se destaca la leucemia linfocítica crónica (LLC) (1), que es una enfermedad neoplásica que se caracteriza por la acumulación de células linfoides de linaje B maduras pero inmunológicamente incompetentes en sangre periférica, médula ósea, ganglios linfáticos, bazo y otros tejidos. Su diagnóstico, en ausencia de infiltración tisular extramedular, requiere una linfocitosis monoclonal sostenida mayor a 5x109/l en sangre periférica, con el inmunofenotipo característico al evaluarla por citometría de flujo (CF) como positividad para CD19, CD20 débil, co-expresión aberrante de CD5, CD23 y expresión débil de inmunoglobulinas de superficie (2).
La causa de la LLC es desconocida; sin embargo, varios estudios coinciden en afirmar que existen factores de riesgo que se relacionan con el desarrollo de la enfermedad como la edad avanzada, sexo masculino y tener antecedentes familiares de esta (3). De hecho, los familiares en primer grado de consanguinidad de pacientes con este tipo de leucemia tienen más de tres veces el riesgo que la población general de padecer esta enfermedad u otra neoplasia linfoide (4) e incluso a edad más temprana con respecto a lo que usualmente se ve en la mayoría de pacientes, lo cual sugiere que hay factores genéticos asociados que influyen en el desarrollo de la misma (3).
En estos parientes y en la población general es posible demostrar mediante citometría de flujo, la presencia en sangre periférica de una población clonal con un inmunofenotipo idéntico al de la LLC; este hallazgo se ha denominado linfocitosis monoclonal de células B (LMB).
La LMB es una condición asintomática que se caracteriza por la circulación en sangre periférica de pequeñas poblaciones clonales de linfocitos B. De acuerdo con las directrices revisadas en el 2008 en el Taller internacional sobre la leucemia linfocítica crónica (por sus siglas en inglés: IWECLL) (2), la LMB supone menos de 5x109/L linfocitos B en la sangre periférica, en ausencia de signos clínicos o síntomas de un trastorno linfoproliferativo crónico de células B (5).
Según la clasificación actual, existen dos tipos de LMB, la tipo LLC y la tipo No-LLC; siendo la más frecuente la tipo LLC (75 % de los casos) (6). Así, en esta última, las células B clonales presentan características inmunofenotípicas similares a las observadas en la LLC ya descritas anteriormente (figura 1).
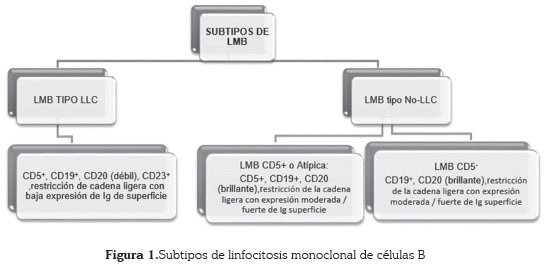
El recuento absoluto de células B es el umbral que permite diferenciar la LMB de la LLC; sin embargo, la progresión de una LMB a una la leucemia que requiere manejo médico ocurre en aproximadamente el 1 % de los casos por año (7). También se ha encontrado que dentro de la LMB el número de células B clonales circulantes puede ser heterogéneo, permitiendo la subclasificación de la LMB tipo LLC en dos subgrupos: una linfocitosis monoclonal de células B clínica (cLMB) y la linfocitosis monoclonal de células B detectada sólo por tamización.
La primera se caracteriza por linfocitosis y una concentración de células B clonales mayor o igual a 1,5x109/L; y la segunda sólo es detectada durante estudios de tamización en población saludable, utilizando técnicas altamente sensibles, y se caracteriza por un recuento de células B clonales menor de 0,05x 109/L, por lo que también es conocida como LMB de recuento bajo (6).
Varios estudios han reportado una prevalencia variable de LMB, dependiente en gran medida de las características de la población examinada (donantes de sangre, adultos mayores de 60 años, familiares de pacientes con LLC, etc.) y de los métodos de detección utilizados para su identificación. Aproximadamente 10 a 15 % de las personas con linfocitosis tienen LMB (8). La prevalencia en el adulto en general (sin incluir aquellos con un historial familiar de LLC) oscila entre 0,12 y 14 % (9).
La ausencia de métodos de laboratorio estandarizados para el diagnóstico de LMB dificulta determinar la verdadera prevalencia, debido a que el uso de métodos más sensibles de citometría de flujo con frecuencia resulta en sobre-estimación del diagnóstico. En estudios realizados en población sana utilizando citometría con dos canales de fluorescencia se detectó una prevalencia de 0,12 a 0,57 % (10,11) y en trabajos en los que se utilizaron cuatro fluorocromos, se informó una prevalencia de un 3,5 a 3,8 % (12,13).
En una investigación realizada en Italia por Fazi et al. (6), utilizando citometría de flujo de cinco colores, se encuentra una prevalencia en personas sanas de 7,7 %. Estas personas tenían recuentos de leucocitos y de linfocitos dentro de los intervalos biológicos de referencia. En un estudio realizado en Salamanca (España) por Nieto et al. (14), que utilizó ocho colores, se informa una prevalencia de LMB tipo LLC de 12 % en 608 sujetos sanos. Cabe resaltar que lo que marcó la diferencia entre estos dos últimos estudios, además del número de fluorescencias, fue el número de células adquiridas, 5x105 células en el estudio italiano y 5x106 en el español, lo que evidencia que la sensibilidad de la técnica aumenta de forma considerable la prevalencia. En este sentido, la utilización de métodos de detección cada vez más sensibles, ha conducido a la identificación de clones cada vez más pequeños de LMB.
Con respecto a los subgrupos de población, la prevalencia más alta de LMB tipo LLC, se ha encontrado en parientes sanos de primer grado de consanguinidad de pacientes con LLC familiar; esta es una condición caracterizada por la presencia de dos o más personas con el diagnóstico de LLC en el interior de una misma familia. En general, la prevalencia de LMB en familiares de pacientes con LLC se ha descrito en un rango que oscila entre el 13,5 y 18 % (15,16).
La LLC es más frecuente en hombres que en mujeres (17) y en LMB también se ha buscado una relación entre la entidad y el sexo. Algunos investigadores han reportado prevalencias mayores en varones (18), aunque la diferencia ha sido menor que en la LLC (19); sin embargo, otros estudios no han mostrado diferencias consistentes en el género (14).
Se ha encontrado que el incremento de la prevalencia de LMB con la edad, tiene un patrón similar al de la LLC. En familias de alto riesgo se ha reportado una prevalencia del 61 % a los 90 años de edad (19) y en la investigación de Nieto et al. (14), en adultos aparentemente sanos, se pudo comprobar que la prevalencia de LMB se incrementa progresivamente con la edad. En el estudio de Matos et al. (18) realizado en familiares de pacientes con LLC esporádica, se observó un comportamiento similar.
Progresión de linfocitosis monoclonal de células B a leucemia linfocítica crónica
Diferentes estudios basados en grandes series hospitalarias de pacientes con LMB (20, 21), han puesto de manifiesto un riesgo más elevado de los sujetos con LMB de progresar a LLC. Un estudio de cohorte prospectivo, realizado por Landgren et al. (21), comprobó la hipótesis que la LLC está siempre precedida por LMB. En dicha investigación se detectaron clones de células B en 44 de 45 muestras (98 %) de linfocitos criopreservados, obtenidos en sangre de sujetos sin cáncer y en los que posteriormente se diagnosticó LLC. Este estado precursor tuvo una duración de seis meses a seis años antes del desarrollo de leucemia.
De hecho, los datos procedentes de trabajos realizados en Reino Unido y Estados Unidos muestran que el recuento absoluto de linfocitos B es el determinante más importante en la progresión a LLC (20, 22); concluyendo que a mayor número de células B clonales, mayor riesgo de desarrollar la enfermedad.
Según la clasificación Rai y Binet, la LLC se categoriza en diferentes estadios que ayudan a definir el pronóstico y la necesidad de instaurar un tratamiento en el paciente y tienen valor principalmente en términos de predicción y de supervivencia; sin embargo, la LMB no hace parte de estos estadios, debido a que es una entidad con un recuento absoluto de linfocitos B menor de 5x109/L.
En este sentido, se han buscado marcadores identificados en pacientes con LLC a través de métodos moleculares, que se han relacionado con agresividad o un curso benigno de la enfermedad. Utilizando hibridación in situ fluorescente (FISH, por sus siglas en inglés) se han podido detectar alteraciones cromosómicas en la LLC, entre las cuales se encuentran deleciones del brazo largo de los cromosoma 13 (23) y 11, delección del brazo corto del cromosoma 17 (24), trisomía 12 y delección del brazo largo del cromosoma 6 (25). Adicionalmente, han encontrado mutaciones somáticas en el gen de la región variable de la cadena pesada de las inmunoglobulinas (IGHV), que han sido relacionadas con un pronóstico favorable (26, 27).
Al estudiar por citometría de flujo, linfocitos B de pacientes con LLC, también se han detectado otros marcadores que han sido relacionados con peor pronóstico de la enfermedad: ZAP-70, CD38 (28,29) y CD49d (30,31). Para LMB también se han realizado estudios que buscan detectar estos factores pronósticos como marcadores de progresión a LLC; dentro de estos se encuentra el realizado por Lanasa et al. (32), en donde la deleción 13q14.3, considerada de pronóstico favorable en LLC, se observó en el 73 % de los casos LMB tipo LLC, siendo un valor más alto que el reportado en publicaciones previas (33); además se reportó el primer caso de deleción 17p13 en LMB de recuento bajo, y debido a que este es un marcador que se encuentra en la LLC que sigue un curso agresivo, se pensaba que rara vez se observa en este tipo de LMB.
En un estudio en el que se realizó un perfil inmunogenético de casos de LMB tipo LLC (con recuentos absolutos altos y bajos de linfocitos B) y se comparó con pacientes con LLC en estadio Rai-0, se comprobó que ambos tipos de LMB tienen estados similares de hipermutación somática; sin embargo, el repertorio de genes IGHV era diferente, al igual que el estereotipo del receptor de célula B (BCR) al compararlos con el de la LLC-0 (34).
Otro reporte en el que se estudiaron 30 marcadores de LLC en LMB, no encuentra diferencias significativas en la expresión de la mayoría de estos, sólo se observan modestas diferencias en la expresión de CD38 y CD49d, ambos casos con pronóstico desfavorable en LLC (35). En un informe se reportó que las personas con una alta proporción de células B clonales CD38+ eran más propensas a desarrollar LLC que requería tratamiento, que aquellos con una baja proporción de estas células (36).
El estudio de Lanasa et al. informa diferencias entre CD38 y CD49d; y sólo el 12 % de las personas con LMB tipo LLC expresaban CD38 en una proporción mayor al 30 % y la expresión de CD49d >45 % sólo se presentó en el 19 % de individuos de este mismo grupo (32). Estas diferencias se hicieron evidentes al comparar estos valores con los reportados previamente para LLC, que fueron de 27 % para CD38 (33) y del 23 % para CD49d (37).
Con respecto a la expresión de ZAP-70, la cual está relacionada con una supervivencia desfavorable en pacientes con LLC, se han encontrado también diferencias significativas: hay reportes de un 26 % de expresión en LMB tipo LLC, en comparación de un 54 % en pacientes con LLC (32).
Algunos investigadores afirman que la mayoría de las personas con LMB tipo LLC no desarrollarán enfermedad progresiva en un período de cinco años aproximadamente, el 75 % estará vivo con un recuento de linfocitos estable y entre el 1 y el 4 % por año desarrollará progresivamente una enfermedad que requiere tratamiento (38). La LMB es por lo menos 100 veces más frecuente que la LLC, lo que indica que sólo un pequeño subconjunto de estos casos, progresa a leucemia (6).
Susceptibilidad genética heredada
Recientemente se han realizado estudios de asociación del genoma completo para identificar polimorfismos de nucleótido simple (SNPs por sus siglas en inglés) que influyen en el riesgo de desarrollar LLC, encontrando seis variantes de bajo riesgo que predisponen a esta enfermedad: 2q13 (rs17483466), 2q37.1(rs13397985), 6p25.3(rs872071), 11q24.1(rs735665), 15q23 (rs7176508) y 19q13.32(rs11083846) (39).
En la LMB también se han estudiado estos SNPs, encontrando frecuencias similares a las documentadas previamente en LLC, que además comprometen genes involucrados en el desarrollo de la enfermedad; por ejemplo rs757978, rs872071 y rs13397985 mapean a genes como FARP2, IRF4, y SP140, respectivamente (8).
Es bien conocido que FARP2 es un importante regulador de la actividad del gen MYC (8), que IRF4 es un regulador clave del desarrollo y proliferación de linfocitos y se ha relacionado con el desarrollo de mieloma múltiple y LLC (39), ya que interactúa con factores de transcripción controlando la terminación de la señalización del receptor de células pre-B y promoviendo la diferenciación de células pro-B a células B pequeñas, además vía Blimp1 (proteína de maduración inducida por linfocitos B) y BCL6 (represor transcripcional requerido para células B del centro germinal), controla la transición de las células B de memoria. En este sentido, SP140 puede ser importante para el establecimiento de infecciones virales latentes y la inmortalización de células B.
Individuos con seis o más alelos tienen un incremento de más de tres veces de riesgo de LMB comparado con los que tienen un número medio de alelos de riesgo (39). Estos datos proporcionan evidencia adicional de que la LMB es una lesión precursora de LLC (8).
Daño temprano en el ADN mediado por ROS y biomarcadores de estrés oxidativo
La LLC se ve afectada por alteraciones en las enzimas antioxidantes y por estrés oxidativo, características comunes de las células tumorales transformadas. El estrés oxidativo juega un papel fundamental en la iniciación y progresión tumoral (40), esto puede ser el resultado de la mayor producción de especies reactivas de oxígeno (ROS), de las cuales se ha propuesto que aumentan la inestabilidad genética a través de sus interacciones con el ADN, conduciendo a la acumulación progresiva de mutaciones genéticas en oncogenes y / o genes supresores de tumores (41).
En los linfocitos B de la LLC el aumento en los productos de oxidación como malondialdehído (MDA) y la base modificada 8-oxo-2’-desoxiguanosina (8-oxo-dG) del ADN, está acompañado por la disminución de las enzimas antioxidantes superóxido dismutasa (SOD) y catalasa (CAT); también se ha reportado aumento en el radio glutatión oxidado / reducido (GSSG/GSH), que es un parámetro indicativo de estrés oxidativo (41).
Basados en los anteriores hallazgos, Collado et al. (40), realizan una investigación para determinar si las alteraciones en el estrés oxidativo descritas en LLC, están presentes en la LMB y para evaluar el rol biológico y clínico del daño oxidativo en estas entidades linfoproliferativas. Para esto estudian 29 pacientes con LMB, 55 pacientes no tratados con LLC y 31 individuos sanos como grupo control. Encuentran que la razón GSSG / GSH, considerada indicativa del estado de óxido-reducción (redox) de las células, fue aproximadamente dos veces mayor en LMB (10,4 %, p < 0,001) y LLC (9,2 %, p < 0,001) que en el grupo control (5,8 %). Por otra parte, la actividad de la catalasa de las células de los grupos de pacientes, fue significativamente inferior que en el grupo control. También se detecta evidencia de actividad de radicales libres en las poblaciones de LMB y LLC en forma de subproductos de la peroxidación de lípidos. Los MDA fueron significativamente mayores en pacientes con LMB (1,2 nmol / mg prot, p <0,001) y LLC (1,1 nmol / mgprot, p <0,001) en comparación con el grupo control (0,4 nmol /mg prot).
Para investigar la ocurrencia de daño en el ADN, se midieron los niveles de 8 -oxo-dG, encontrando una frecuencia de bases dañadas en pacientes con LMB y LLC ocho veces mayor que en los sujetos sanos. El aumento en los marcadores de estrés oxidativo, junto con la disminución de la actividad antioxidante de la catalasa, son indicativos de la vulnerabilidad de las células LMB y LLC a la peroxidación de lípidos y el daño en el ADN. (40).
Estudio de linfocitosis monoclonal de células B en médula ósea
La información acerca de la presencia de LMB tipo LLC en médula ósea es muy escasa y generalmente suele ser detectada como un hallazgo incidental en muestras tomadas para la investigación de linfoma. Sin embargo, mediante el empleo de técnicas sensibles de identificación es probable acercarse a la prevalencia vista en la población general de hasta el 5 % (42).
La presencia de una pequeña población de células con fenotipo de LLC no debe ser considerada como evidencia de compromiso de la médula ósea, a menos que pueda demostrarse una relación de clonalidad tanto en estas células como en las de la biopsia de tejido. No se debe suponer que la detección de LMB tipo LLC en niveles bajos en sangre periférica o médula ósea descarta la necesidad de la biopsia de ganglio linfático en individuos sometidos a evaluación de linfadenopatía.
Así mismo, en un individuo que presenta citopenia la identificación de LMB tipo LLC no elimina la necesidad de una investigación mayor, para determinar la causa subyacente de la citopenia; menos del 5 % de los individuos sin linfadenopatía y con menos de 5x109/L células de LLC circulantes tienen compromiso extenso de la médula ósea relacionado con LLC (42).
En la mayoría de las personas con LMB tipo LLC y que requieren un aspirado de médula ósea para la investigación de citopenia subyacente, la causa generalmente es otra enfermedad, como un carcinoma metastásico o un síndrome mielodisplásico, que no está relacionado con LLC (42).
Tamización de linfocitosis monoclonal de células B en donantes de progenitores hematopoyéticos
El hallazgo de LMB en estudios realizados en la población general y el aumento de la prevalencia en familias con historia de LLC plantea una nueva dificultad en la realización de trasplantes de progenitores hematopoyéticos, en especial si el donante es un familiar en primer grado de consanguinidad del receptor, y si este último ha sido diagnosticado con LLC.
Recientemente se han reportado algunos casos de transmisión de células leucémicas por trasplante alogénico de células madre proveniente de familiares de pacientes con LLC. Flandrin-Gresta et al. (43), reportan un caso de transmisión de LLC por trasplante de células madre de sangre periférica ligado a la presencia de LMB en el donante (hermano HLA compatible); hallazgo que ha llevado a proponer la utilización de métodos sensibles para la detección de esta entidad en los donantes, particularmente cuando existe una historia de LLC en la familia.
Herishanu et al. (44), reportaron un caso de un donante de células madre de sangre periférica, al cual se le diagnosticó LMB en el momento del trasplante. Ellos afirman que hasta ahora, el riesgo potencial de transferencia de LMB de donante a receptor a través de trasplante alogénico de células madre es desconocido. Obviamente, la principal preocupación en estos casos es la transferencia del clon de LMB para el receptor, el cual potencialmente puede progresar de forma más agresiva por la inmunosupresión asociada con el régimen de preparación para el trasplante.
Diagnóstico diferencial y manejo clínico de la linfocitosis monoclonal de células B
El diagnóstico diferencial entre LMB, LLC y el linfoma linfocítico de célula pequeña, se basa en el recuento de células B en sangre periférica y en los exámenes físicos de los pacientes.
La LMB se caracteriza por un recuento absoluto de células B menor de 5x109células/L y la ausencia de linfadenopatía y hepatoesplenomegalia; por su parte, el linfoma linfocítico de célula pequeña también se caracteriza por un recuento absoluto de células B menor de 5x109 células/L pero se presenta un aumento en el tamaño de ganglios linfáticos, del hígado o del bazo (45). En cuanto a la LLC, se define por la presencia de más de 5x109 células B/L en sangre periférica, y la linfadenopatía y hepatoesplenomegalia se encuentran en diversos grados dependiendo el estadio de la enfermedad (46).
El manejo clínico de los individuos con LMB es diferente según el subtipo característico de la entidad. En términos prácticos, el médico general podría hacer seguimiento a las personas con LMB de recuento bajo, dado que estos individuos tienen hemograma completamente normal y, en particular la experiencia clínica ha demostrado que la progresión en este grupo es muy rara. Para esas personas, no es necesario realizar una estrecha vigilancia y es suficiente y apropiado un examen anual con un recuento sanguíneo completo (46).
Tal como se referenció anteriormente, los pacientes con cLMB tienen un riesgo de progresión a una LLC que requiere tratamiento de 1 % por año (7). Basados en este bajo riesgo de progresión se recomienda un seguimiento anual realizando un cuadro hemático completo. Los pacientes deben estar atentos a síntomas específicos, tales como aumento de tamaño de los ganglios linfáticos, sudoración nocturna, fatiga extrema y pérdida de peso, dado que estos hallazgos clínicos podría ser la primera evidencia de progresión de la enfermedad (46).
Los pacientes con LMB atípica o CD5 negativa requieren una evaluación diferente; en el caso de existir manifestaciones clínicas se debe proceder a realizar una evaluación médica y de laboratorio completa que permita la clasificación específica del subtipo de linfoma no Hodgkin (46).
CONCLUSIONES
Existe gran variedad en la prevalencia de LMB, dependiente en gran medida de las características de la población examinada y de los métodos de detección utilizados para su identificación.
La LMB, es una categoría de riesgo diferente y más favorable que LLC Rai 0. Los pacientes con cLMB deben someterse a un seguimiento regular y deben ser evaluados para uno o más de los factores biológicos de riesgo disponibles.
Los factores de riesgo que determinan el paso de LMB a LLC están aún por determinarse, sin embargo diversos estudios han reportado que la edad, el sexo, los antecedentes familiares de LLC, la presencia de algunos polimorfismos de nucleótido simple (SNPs) y marcadores como ZAP 70, CD38 y CD49d, podrían estar relacionados con el riesgo de desarrollar la enfermedad. Sin embargo hasta ahora el factor mejor definido de progresión es el recuento absoluto de linfocitos B.
CONFLICTO DE INTERESES
No existe ninguno.
BIBLIOGRAFÍA
1. Gómez MA, Tarín LC, Cantú O, Gutiérrez CH, Méndez N, Gómez D. La leucemia linfocítica crónica no es la única causa de linfocitosis persistente. Medicina Universitaria. 2008;10(41):212-5. [ Links ]
2. Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Dohner H, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the InternationalWorkshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111(12):5446-56. [ Links ]
3. Goldin LR, Slager SL, Caporaso NE. Familial chronic lymphocytic leukemia. Curr Opin Hematol. 2010;17(4):350-5. [ Links ]
4. Beutler E, Coller BS, Kipps TJ, Seligsohn U. Williams Hematología. 6 ed. Madrid: Mc Graw Hill; 2005. [ Links ]
5. Marti GE, Rawstron AC, Ghia P, Hillmen P, Houlston RS, Kay N, et al. Diagnostic criteria for monoclonal B-cell lymphocytosis. Br J Haematol. 2005;130(3):325-32. [ Links ]
6. Fazi C, Scarfo L, Pecciarini L, Cottini F, Dagklis A, Janus A, et al. General population low-count CLL-like MBL persists over time without clinical progression, although carrying the same cytogenetic abnormalities of CLL. Blood. 2011;118(25):6618-25. [ Links ]
7. Shanafelt TD, Kay NE, Rabe KG, Call TG, Zent CS, Maddocks K, et al. Brief report: natural history of individuals with clinically recognized monoclonal B-cell lymphocytosis compared with patients with Rai 0 chronic lymphocytic leukemia. J Clin Oncol. 2009;27(24):3959-63. [ Links ]
8. Crowther-Swanepoel D, Corre T, Lloyd A, Gaidano G, Olver B, Bennett FL, et al. Inherited genetic susceptibility to monoclonal B-cell lymphocytosis. Blood. 2010;116(26):5957-60. [ Links ]
9. Shim YK, Middleton DC, Caporaso NE, Rachel JM, Landgren O, Abbasi F, et al. Prevalence of monoclonal B-cell lymphocytosis: A systematic review. Cytometry Part B - Clinical Cytometry. 2010;78(SUPPL. 1):S10-S8. [ Links ]
10. Rachel JM, Zucker ML, Fox CM, Plapp FV, Menitove JE, Abbasi F, et al. Monoclonal B-cell lymphocytosis in blood donors. British Journal of Haematology. 2007;139(5):832-6. [ Links ]
11. Shim YK, Vogt RF, Middleton D, Abbasi F, Slade B, Lee KY, et al. Prevalence and natural history of monoclonal and polyclonal B-cell lymphocytosis in a residential adult population. Cytometry Part B - Clinical Cytometry. 2007;72(5):344-53. [ Links ]
12. Rawstron AC, Green MJ, Kuzmicki A, Kennedy B, Fenton J, Evans P et al. Monoclonal B lymphocytes with the characteristics of "in dolent" chronic lymphocytic leukemia are present in 3.5 % of adults with normal blood counts. Blood. 2002;100(2):635-9. [ Links ]
13. Ghia P, Prato G, Scielzo C, Stella S, Geuna M, Guida G, et al. Monoclonal CD5+ and CD5-B-lymphocyte expansions are frequent in the peripheral blood of the elderly. Blood. 2004;103(6):2337-42. [ Links ]
14. Nieto WG, Almeida J, Romero A, Teodosio C, Lopez A, Henriques AF, et al. Increased frequency (12 %) of circulating chronic lymphocytic leukemia-like B-cell clones in healthy subjects using a highly sensitive multicolor flow cytometry approach. Blood. 2009;114(1):33-7. [ Links ]
15. Rawstron AC, Yuille MR, Fuller J, Cullen M, Kennedy B, Richards S, et al. Inherited predisposition to CLL is detectable as subclinical monoclonal B-lymphocyte expansion. Blood. 2002;100(7):2289-90. [ Links ]
16. Marti GE, Carter P, Abbasi F, Washington GC, Jain N, Zenger VE, et al. B-Cell Monoclonal Lymphocytosis and B-Cell abnormalities in the setting of familial B-Cell Chronic Lymphocytic Leukemia. Cytometry Part B - Clinical Cytometry. 2003;52(1):1-12. [ Links ]
17. Weber W, Maurer PF, Estoppey J, Zwahlen M. Chronic lymphocytic leukaemia: clinical-aetiological findings in 66 patients and their families. Hered Cancer Clin Pract. 2007;5(4):210-2. [ Links ]
18. Matos DM, Ismael SJ, Scrideli CA, de Oliveira FM, Rego EM, Falcao RP. Monoclonal B-cell lymphocytosis in first-degree relatives of patients with sporadic (non-familial) chronic lymphocytic leukaemia. Br J Haematol. 2009;147(3):339-46. [ Links ]
19. Goldin LR, Lanasa MC, Slager SL, Cerhan JR, Vachon CM, Strom SS, et al. Common occurrence of monoclonal B-cell lymphocytosis among members of high-risk CLL families. Br J Haematol. 2010;151(2):152-8. [ Links ]
20. Rawstron AC, Bennett FL, O'Connor SJ, Kwok M, Fenton JA, Plummer M, et al. Monoclonal B-cell lymphocytosis and chronic lymphocytic leukemia. N Engl J Med. 2008;359(6):575-83. [ Links ]
21. Landgren O, Albitar M, Ma W, Abbasi F, Hayes RB, Ghia P, et al. B-cell clones as early markers for chronic lymphocytic leukemia. N Engl J Med. 2009;360(7):659-67. [ Links ] .
22. Shanafelt TD, Kay NE, Jenkins G, Call TG, Zent CS, Jelinek DF, et al. B-cell count and survival: differentiating chronic lymphocytic leukemia from monoclonal B-cell lymphocytosis based on clinical outcome. Blood. 2009;113(18):4188-96. [ Links ]
23. Rodrıguez AE, Hernandez JA, Benito R, Gutierrez NC, Garcıa JL, Hernandez-Sanchez M. Molecular Characterization of Chronic Lymphocytic Leukemia patients with a high number of losses in 13q14. PLoS One. 2012;7(11):1-13. [ Links ]
24. Gladstone DE, Blackford A, Cho E, L S, Kasamon Y, Gocke C. The Importance of IGHV Mutational Status in del(11q) and del(17p) Chronic Lymphocytic Leukemia. Clin Lymphoma Myeloma Leuk. 2012; 12(2):132-7. [ Links ]
25. Gunnarsson R, Mansouri L, Isaksson A, Göransson H, Cahill N, Jansson M, et al. Array-based genomic screening at diagnosis and during follow-up in chronic lymphocytic leukemia. Haematologica. 2011;96(8):1161-9. [ Links ]
26. Damle RN, Wasil T, Fais F, Ghiotto F, Valetto A, Allen SL, et al. Ig V gene mutation status and CD38 expression As novel prognostic indicators in chronic lymphocytic leukemia. Blood. 1999;94(6):1840-7. [ Links ]
27. Sagatys EM, Zhang L. Clinical and laboratory prognostic indicators in chronic lymphocytic leukemia. Cancer Control. 2012;19(1):18-25. [ Links ]
28. Grever MR, Lucas DM, Dewald GW, Neuberg DS, Reed JC, Kitada S, et al. Comprehensive assessment of genetic and molecular features predicting outcome in patients with chronic lymphocytic leukemia: results from the US Intergroup Phase III Trial E2997. J Clin Oncol 2007;25(7): 799-804. [ Links ]
29. Teimori H, Akbari MT, Hamid M, Forouzandeh M, Bibordi E. Analysis of CD38 and ZAP70 mRNA expression among cytogenetic subgroups of Iranian chronic-lymphocytic-leukemia patients Genet Mol Res. 2011;10(4):2415-23. [ Links ]
30. Gattei V, Bulian P, Del Principe MI, Zucchetto A, Maurillo L, Buccisano F, et al. Relevance of CD49d protein expression as overall survival and progressive disease prognosticator in chronic lymphocytic leukemia. Blood. 2008;111(2):865-73. [ Links ]
31. Buggins AG, Pepper C, Patten PE, Hewamana S, Gohil S, Moorhead J, et al. Interaction with vascular endothelium enhances survival in primary chronic lymphocytic leukemia cells via NF-kappaB activation and de novo gene transcription. Cancer Res. 2010;70(19):7523-33. [ Links ]
32. Lanasa MC, Allgood SD, Slager SL, Dave SS, Love C, Marti GE, et al. Immunophenotypic and gene expression analysis of monoclonal B-cell lymphocytosis shows biologic characteristics associated with good prognosis CLL. Leukemia. 2011;25(9):1459-66. [ Links ]
33. Weinberg JB, Volkheimer AD, Chen Y, Beasley BE, Jiang N, Lanasa MC, et al. Clinical and molecular predictors of disease severity and survival in chronic lymphocytic leukemia. Am J Hematol. 2007;82(12):1063-70. [ Links ]
34. Vardi A, Dagklis A, Scarfò L, Jelinek D, Newton D, Bennett F, et al. Immunogenetics shows that not all MBL are equal: the larger the clone, the more similar to CLL. Blood. 2013 121(22):4521-8. [ Links ]
35. Rawstron AC, Shingles J, de Tute R, Bennett F, Jack AS, Hillmen P. Chronic lymphocytic leukaemia (CLL) and CLL-type monoclonal B-cell lymphocytosis (MBL) show differential expression of molecules involved in lymphoid tissue homing. Cytometry B Clin Cytom. 2010;78 Suppl 1:S42-6. [ Links ]
36. Rossi D, Sozzi E, Puma A, De Paoli L, Rasi S, Spina V, et al. The prognosis of clinical monoclonal B cell lymphocytosis differs from prognosis of Rai 0 chronic lymphocytic leukaemia and is recapitulated by biological risk factors. Br J Haematol. 2009;146(1):64-75. [ Links ]
37. Shanafelt TD, Geyer SM, Bone ND, Tschumper RC, Witzig TE, Nowakowski GS, et al. CD49d expression is an independent predictor of overall survival in patients with chronic lymphocytic leukaemia: a prognostic parameter with therapeutic potential. Br J Haematol. 2008;140(5):537-46. [ Links ]
38. Rawstron AC, Hillmen P. Clinical and diagnostic implications of monoclonal B-cell lymphocytosis. Best Practice & Research Clinical Haematology. 2010;23(1):61-9. [ Links ]
39. Di Bernardo MC, Crowther-Swanepoel D, Broderick P, Webb E, Sellick G, Wild R, et al. A genome-wide association study identifies six susceptibility loci for chronic lymphocytic leukemia. Nat Genet. 2008;40(10):1204-10. [ Links ]
40. Collado R, Oliver I, Tormos C, Egea M, Miguel A, Cerda C, et al. Early ROS-mediated DNA damage and oxidative stress biomarkers in Monoclonal B Lymphocytosis. Cancer Lett. 2012;317(2):144-9. [ Links ]
41. Oltra AM, Carbonell F, Tormos C, Iradi A, Sáez GT. Antioxidant enzyme activities and the production of MDA and 8-oxo-dG in chronic lymphocytic leukemia. Free Radic Biol Med. 2001;30(11):1286-92. [ Links ]
42. Rawstron AC. Monoclonal B-cell lymphocytosis. Hematology Am Soc Hematol Educ Program. 2009:430-9. [ Links ]
43. Flandrin-Gresta P, Callanan M, Nadal N, Jaubert J, Cornillon J, Guyotat D, et al. Transmission of leukemic donor cells by allogeneic stem cell transplantation in a context of familial CLL: should we screen donors for MBL? Blood. 2010; 116( 23):5077- 8. [ Links ]
44. Herishanu Y, Eshel R, Kay S, Rothman R, Njuguna N, Perry C, et al. Unexpected detection of monoclonal B-cell lymphocytosis in a HLA-matched sibling donor on the day of allogeneic stem cell transplantation for a patient with chronic lymphocytic leukaemia: clinical outcome. Br J Haematol. 2010;149(6):905-7. [ Links ]
45. Matos DM, Falcao RP. Monoclonal B-cell lymphocytosis: a brief review for general clinicians. Sao Paulo Med J. 2011;129(3):171-5. [ Links ]
46. Shanafelt TD, Ghia P, Lanasa MC, Landgren O, Rawstron AC. Monoclonal B-cell lymphocytosis (MBL): biology, natural history and clinical management. Leukemia. 2010;24(3):512-20. [ Links ]
