











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
La osteogénesis imperfecta es un grupo de anomalías genéticas del colágeno tipo 1, principalmente de los genes COL1A1 y COL1A2, que afectan tanto la producción como la estructura del colágeno tipo I, generando diferentes déficits de masa ósea y una susceptibilidad aumentada a las fracturas 1,2. La prevalencia estimada varía desde 0,5 a 1 en cada 10 000 habitantes 3.
La clasificación de la enfermedad ha variado en el tiempo: la primera de ellas y sobre la que se basan las actualizaciones de la nomenclatura es la de Sillence et al. de 1979 2. Originalmente, definía cuatro tipos, de acuerdo con la severidad del fenotipo y los patrones de herencia, así: tipo I o leve, acompañado de escleras azuladas con herencia dominante; tipo II, letal en el periodo perinatal con fémur curvo y costillas con nódulos; tipo III con fracturas y deformidad progresiva; y, tipo IV con herencia dominante y escle ras normales. Algunos de los tipos II y III, presentan herencia autosómica recesiva y heterogeneidad genética. Se han hecho varias propuestas para la actualización de la nomenclatura involucrando las aliteraciones genéticas al fenotipo, como la de Van Dijk y Sillence, en 2014 1.
La osteogénesis imperfecta tipo IV es la menos frecuente y es la segunda con menor severidad en el fenotipo. Los afectados presentan baja estatura, múltiples fracturas de severidad variable, escoliosis, dentinogénesis imperfecta, escleras blancas o grisáceas y sólo el 10 % de los pacientes presentan escleras azul pálido 4. En este tipo de la enfermedad se ha demostrado que el manejo integral disminuye el número de fracturas y mejora la calidad de vida 5.
En casos de fetos con identificación prenatal de huesos largos cortos debe incluirse en el diagnóstico etiológico los tipos de osteogénesis imperfecta no letales y no relacionados con fracturas intrauterinas, esto con el objetivo de que se realice un seguimiento ecográfico en útero, y en los nacidos clínico y radiológico, con lo que se realizarían intervenciones tempranas que prevendrían fracturas.
La divulgación de hallazgos de asociaciones de una enfermedad con variantes ge néticas específicas, además de ser de interés científico, es información útil para la interpretación de resultados de pruebas de secuenciación. Reportamos un caso colombiano de osteogénesis imperfecta tipo IV, cuyo diagnóstico se realizó mediante pruebas moleculares, y se brinda información sobre la clínica, métodos diagnósticos y las posibilidades terapéuticas.
Descripción del caso
Se trataba de una paciente de seis meses de edad al momento de la consulta, pro ducto de unión de padres no consanguíneos. Madre de 24 años, quien hizo adecuado control prenatal y no presentaba enfermedades relevantes, ni reportaba exposiciones teratogénicas. Le realizaron tres ecografías prenatales y en la última, a las 34 semanas, reportaron huesos largos por debajo del percentil 5. La niña nació por parto a las 40 semanas de gestación, sin complicaciones con peso de 3140 g y estatura de 48 cm.
A los dos meses la madre notó deformidad en fémur por lo que consultó con médico general, quien solicitó radiografías y remitió a ortopedia pediátrica, en donde encon traron fémures cortos, escleras azules y sospecharon osteogénesis imperfecta.
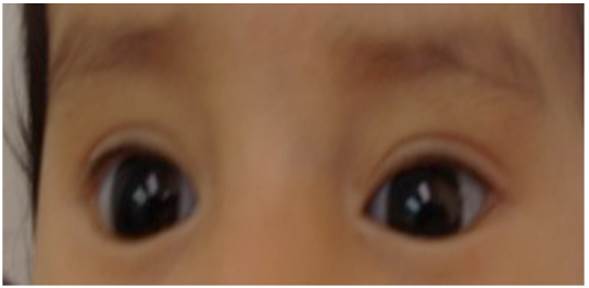
A los dos meses y 20 días de edad fue valorada por genética y dismorfología encon trando peso 4 690 g (percentil 25-50), talla 56 cm (percentil 50), perímetro cefálico de 37,3 cm (percentil 25-50) y hallazgos al examen físico como cara triangular, es cleras azuladas, fisuras palpebrales inclinadas hacia arriba (Figura 1); labio superior delgado en tienda de campaña, acortamiento del fémur bilateral y desviación en varo de la tibia. No se reportaban antecedentes familiares de osteogénesis imperfecta.
Traía resultados de laboratorios así: hormona estimulante del tiroides 4,0 μUI/ml, lactato deshidrogenasa 653 U/L y fosfatasa alcalina 367 U/L, todos dentro de rangos de normalidad.
Una ecografía abdominal total fue reportada como normal. El ecocardiograma evi denció pequeña comunicación interauricular tipo ostium secundum, comunicación interventricular apical; ambos hallazgos sin repercusión hemodinámica y en re visión por cardiología pediátrica. Se le realizaron potenciales evocados visuales y auditivos, los cuales fueron normales.
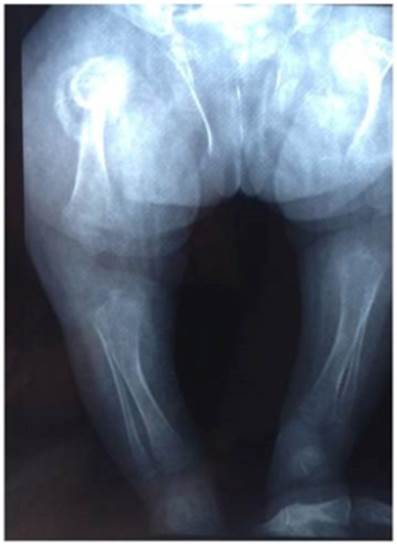
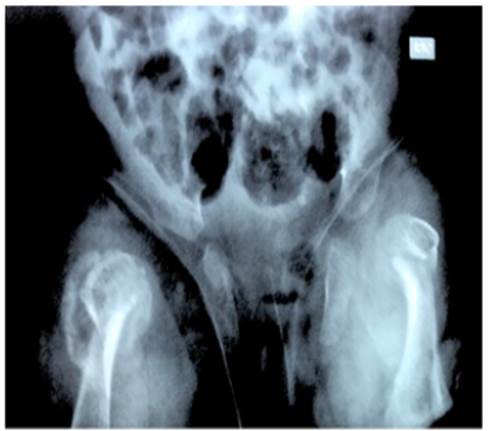
Una radiografía de cráneo evidenció desproporción craneofacial y huesos wormianos. La radiografía de la región cervical indicó fusión atlanto-odontoidea y otra de cadera y miembros inferiores, displasia bilateral de cadera. Evidenciándose que los huesos de los miembros inferiores eran cortos y presentaban angulación lateral. Estos ha llazgos se visualizaban especialmente a nivel femoral. La cortical estaba adelgazada y se observaba ensanchamiento de las metáfisis. Existía una fractura antigua con formación de callo óseo en la región proximal del fémur derecho. Igualmente, se ob servó displasia bilateral de cadera y disminución generalizada de la densidad ósea (Figuras 2 y 3).
Se consideró que los hallazgos eran compatibles con osteogénesis imperfecta a clasificar, teniendo como principales posibilidades los tipos I y IV, dado que la de tipo II es letal y el tipo III es severamente deformante. Se solicitó panel molecular para osteogénesis imperfecta que incluyó los genes ALPL, COL1A1, COL1A2 e IFITM5.
Tres meses después la madre y la paciente regresaron a consulta de genética y dis morfología. Con seis meses de edad se encontró un peso 6 340 g (percentil 10-25), talla 61 cm (percentil 3), perímetro cefálico de 40 cm (percentil 3-10). Mostrando una disminución importante en el percentil de la talla.
No se encontraron otros hallazgos relevantes en el examen físico o nuevas fracturas diferentes a los de la consulta inicial. Se realizó un panel molecular mediante secuen ciación masiva (NGS, del inglés: next generation sequencing), utilizando la tecnología Ilumina, amplificando los exones de los genes ALPL, COL1A1, COL1A2 e IFITM5. Se de tectó en el gen COL1A2 una transición en heterocigosis de una guanina a una adenina (c.2531G>A) que da lugar, a nivel de la proteína, al cambio de la glicina de la posición 844 por un ácido aspártico (p.Gly844Asp), LGR: NG_007405.1.
Dado que los hallazgos fenotípicos sugerían las formas de presentación moderada de osteogénesis imperfecta, compatibles con la vida extrauterina, que se detectó variante en el gen COL1A2 en heterocigosis el cual no se encuentra afectado en la osteogénesis imperfecta tipo I y sí en la tipo IV, que adicionalmente los demás tipos descritos en los últimos años no presentan alteraciones en los genes COL1A1 ni COL1A2, y que los padres no presentaban hallazgos sugestivos de osteogénesis imperfecta, se concluyó que la paciente tenía osteogénesis imperfecta tipo IV, autosómica dominante, secun daria a una variante de novo.
Se realizó asesoría genética a los padres, se consideró que no necesitaban estudios moleculares del gen COL1A2, dado que el patrón de expresión de la variante encon trada en la paciente era dominante y se les explicó que era improbable que ellos fueran portadores de la misma variante. Así mismo, se les informó sobre la baja probabilidad de tener otro hijo con la enfermedad y que la niña cuando estuviera en edad reproductiva tendría en cada embarazo un 50 % de riesgo de tener un hijo con osteogénesis imperfecta tipo IV.
Se remitió la paciente a endocrinología, pediatría y ortopedia con la propuesta tera péutica de iniciar bifosfonatos y terapia física.
Discusión
La osteogénesis imperfecta representa un grupo de anomalías genéticas de caracte rísticas clínicas heterogéneas. Los pacientes varían desde asintomáticos en edades tempranas, estatura normal y fracturas leves hasta manifestaciones graves como muerte perinatal, deformidades esqueléticas, fracturas múltiples y déficit en el desa rrollo motor 1,2,4.
En el 90 % de los casos la osteogénesis imperfecta corresponde a una variante autosómica dominante en los genes COL1A1 o COL1A2, generando anomalías en la síntesis o la estructura del colágeno tipo I. Las fibras de colágeno tipo I contribuyen a la ductilidad y la resistencia del hueso, a diferencia de la rigidez y la fuerza que vienen dadas principalmente por el componente mineral. El colágeno tipo I se produce inicialmente como procolágeno tipo I constituido por dos cadenas proa1 y una proa2, las cuales están codificados por los genes COL1A1 y COL1A2, respectivamente 3. La paciente tenía una variante en el gen COL1A2 en heterocigosis.
Cada cadena de procolágeno se compone, en gran parte, de una secuencia repetitiva de aminoácidos en el orden glicina-X-Y. Las cadenas se alinean y sufren una modi ficación postraduccional en el retículo endoplásmico rugoso (rER) combinándose en una triple hélice. Para que las tres cadenas se entrelacen correctamente deben tener un residuo de glicina en cada tercera posición. Un complejo en el rER com puesto por la prolil 3-hidroxilasa (P3H1), la proteína asociada a cartílago (CRTAP) y la ciclofilina B (CypB), facilitan tanto el plegamiento eficiente de la triple hélice como la 3-hidroxilación de una sola prolina en la cadena alfa1 1,3.
Las anormalidades más comunes asociadas con osteogénesis imperfecta son va riantes puntuales que afectan a un residuo de glicina en alguno de los genes COL1A1 o COL1A27. Así, las células con la alteración producen una mezcla de colágeno normal y anormal que genera huesos frágiles altamente susceptibles a las frac turas, ligamentos laxos y demás características de la enfermedad. El fenotipo re sultante puede variar de leve a letal, dependiendo de cuál de las dos cadenas alfa es afectada, en qué posición de la triple hélice se produce la sustitución y por cuál aminoácido es sustituida la glicina 3.
De acuerdo a la clasificación de Sillence 2, la enfermedad tipo I es la forma de pre sentación más leve y tiene una prevalencia de 3 a 5 casos por 100 000 nacimientos; la tipo II, que corresponde a la forma de presentación más grave, es congénita, tiene un alto índice de muerte perinatal y una prevalencia de 1 caso por 40 000 a 60 000 nacimientos; la tipo III, que se presenta con deformidades severas, corresponde a la segunda en gravedad y tiene una prevalencia de 1 a 2 casos por 100 000 naci mientos; y la tipo IV, la otra forma de presentación leve a moderada y que hasta el momento no tiene una prevalencia establecida, existiendo solamente algunos casos reportados 1,8,9.
Se han descrito otros tipos que abarcan variantes en otros genes, pero aún no se cuenta con datos epidemiológicos sobre la frecuencia de su presentación 1,10.
Warman et al., en el 2011, después de la reunión del Grupo Internacional de Nomen clatura de Desórdenes Esqueléticos, incluyen la tipo V, con calcificaciones de las membranas interóseas e hipertrofia del callo 11.
Van Dijk y Sillence en el 2014 proponen una nueva nomenclatura según los genes afectados y el fenotipo. Clasifican en el grupo A los pacientes con fenotipo de leve a moderada severidad y en el B a quienes tienen un fenotipo progresivamente defor mante y muerte perinatal. En el grupo A estarían incluidos la osteogénesis imperfecta tipos I y IV, y en el B los tipos II, III y V 1.
El caso reportado corresponde a una paciente con una osteogénesis imperfecta tipo IV, según Sellience y del grupo A según Van Dijk, una forma de presentación leve a moderada, donde los hallazgos clínicos que sustentaron el diagnóstico fueron: acor tamiento y angulación del fémur bilateral, fractura de fémur derecho, desviación en varo de la tibia, luxación coxofemoral bilateral, huesos wormianos, osteopenia y talla baja. Una particularidad de este caso son las escleras azuladas, hallazgo que se ha encontrado solamente en el 10 % de los pacientes con osteogénesis imperfecta tipo IV, presentándose la mayoría con escleras normales o grisáceas 12.
El tipo de herencia en la osteogénesis imperfecta tipo IV es autosómico dominante y los genes afectados pueden ser el COL1A1 o el COL1A212; también se han re portado algunas familias con patrón de herencia recesiva secundario a variantes en los genes CRTAP, PPIB, SP7 en homocigosis 1. En este caso, se evidenció en el gen COL1A2 una transición en heterocigosis de una guanina a una adenina (c.2531G>A) que da lugar, a nivel de la proteína, al cambio de la glicina de la posición 844 por un ácido aspártico (p.Gly844Asp).
Teniendo en cuenta que el gen afectado fue el COL1A2 y que la presentación clínica es compatible con la vida y con una deformidad esquelética moderada, se pueden descartar las formas de presentación tipo I (gen afectado COL1A1), tipo II (letal), III (severamente deformante) y otros tipos de osteogénesis imperfecta descritos que presentan variantes en genes diferentes a los que codifican las cadenas del colágeno tipo I 13,14,15,16.
La variante génica específica descrita ya había sido reportada en 2006, concomitan temente en una madre y su hija, ambas afectadas con osteogénesis imperfecta. La madre, de 28 años, había presentado múltiples fracturas, escleras azules, sin otros hallazgos y la recién nacida presentó fémures cortos y arqueados sin fracturas. Los autores interpretan el fenotipo como osteogénesis imperfecta tipo I, aunque recono cieron que las variantes en el gen COL1A2 eran relacionadas con el tipo IV 6.
La base de datos Clinvar del National Center for Biotechnology Information (NCBI) reporta como probablemente patogénica una variante de cambio de sentido del mismo nucleótido 17. Esto sugiere que cambios del aminoácido 844 resultan en una alte ración del papel fisiológico de esta proteína.
El tratamiento farmacológico presenta varias opciones, siendo los bifosfonatos los medicamentos más utilizados 5,9,18. Estos fármacos actúan inhibiendo los os teoclastos y, por ende, inhibiendo la resorción ósea y aumentando la densidad ósea. Un estudio reciente en el que utilizan pamidronato de sodio, que incluye 23 pacientes con osteogénesis imperfecta tipo IV, reporta que es seguro, bien tolerado y muestra mejoría en la densidad ósea y reducción de fracturas en los pacientes adolescentes 19. Sin embargo, los bifosfonatos presentan un mayor pico de acción entre los dos y los cuatro años y, a pesar de los beneficios documentados y su uso extendido du rante los últimos años, aún se desconoce la dosis óptima, el tiempo de duración del tratamiento y los efectos adversos a largo plazo 15.
Debido a sus potenciales efectos anabólicos en el hueso a través de la estimulación de los osteoblastos, la síntesis de colágeno y el crecimiento óseo, se ha probado la hormona de crecimiento en ensayos clínicos. Se ha visto que su uso, solo o en compañía de bifosfonatos, aumenta la densidad ósea aunque no reduce la tasa de fracturas y faltan estudios para indicar el uso estandarizado en esta enfermedad 5.
Existen terapias en estudio como los inhibidores del receptor del activador del ligando del factor nuclear-kB, como denosumab, que inhiben la formación de osteoclastos y la degradación de hueso, los anticuerpos anti Sclerostin y Dickkopf-1 que aumentan la actividad de los osteoblastos y la formación de hueso perióstico a través de la inhibición de la vía de Wnt, pero aun se requieren más estudios para implementar su uso. También se han intentado autotransplantes en médula ósea, de células sin la variante en los genes que modifican el colágeno, pero aún no hay resultados con cluyentes 5.
La rehabilitación física está orientada a la adquisición de habilidades motoras, dis minución del tiempo de quietud secundario a las fracturas y ganancia de masa mus cular. La cirugía se utiliza para tratar de disminuir el tiempo de recuperación de las fracturas, la osteopenia y la debilidad muscular por desuso que pueden inducir a más fracturas y, adicionalmente, intenta reducir o corregir las deformidades dadas por estas 5,14. La movilidad de los afectados mejora cuando reciben bifosfonatos y realizan terapias físicas especificas 20.
El manejo debe ser multidiciplinario e incluye rehabilitación física, cirugía ortopé dica, tratamiento farmacológico, control por pediatría y endocrinología pediátrica y asesoría genética. El grado de intervención de cada paciente está dado por la seve ridad del fenotipo clínico y se orienta a maximizar la movilidad, mejorar las compe tencias de la vida diaria y disminuir el dolor y la fragilidad ósea. Además de explicar los riesgos de recurrencia a la familia 5,9,14.
Conclusión
Se reporta un caso de osteogénesis imperfecta tipo IV con diagnóstico clínico y mole cular, con una rara variante de cambio de sentido en COL1A2 (c.2531G>A).
Además de aportar información a la comunidad médica para generar diagnósticos tempranos y una clasificación adecuada según el fenotipo y el genotipo que permita brindar un manejo integral oportuno que mejore el pronóstico y realizar asesoría genética.

