











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
La inmunodeficiencia más común en humanos es la deficiencia de IgA 1, con una prevalencia ampliamente variable en diferentes poblaciones 2-4; puede ser asintomática o sintomática 5-7 y suele ser incluso un hallazgo incidental durante la cuantificación de las inmunoglobulinas séricas 8-10. Para diagnosticarla, se cuantifica la concentración de IgA en sangre y se evalúa la magnitud de su disminución. De acuerdo con esta evaluación se clasifica en deficiencia parcial (DPIgA) o deficiencia total (DTIgA). Adicionalmente, si solo se afectan los niveles de IgA sin alteraciones de otras inmunoglobulinas séricas como IgM e IgG o subclases de inmunoglobulina G, entonces se denomina como deficiencia selectiva de IgA (DSIgA). La deficiencia de IgA puede ser primaria y su origen es desconocido, o secundaria que suele deberse a infecciones o a el consumo de ciertos medicamentos 11. En la mayor parte de los casos esta deficiencia es reversible, aunque la recuperación total de los niveles séricos de IgA puede tardar meses o años 12. Se ha reportado la asociación de infecciones con una disminución drástica de los niveles de IgA; sin embargo, no se demuestra una relación causa-efecto, en cuanto no se reportan dosificaciones antes de la infección, ni se hace un seguimiento prospectivo de estos valores 13,14.
El propósito de la presente investigación es presentar un nuevo algoritmo clínico que nos permita mejorar la sospecha clínica, el diagnóstico y brindar un adecuado manejo clínico de los pacientes con esta inmunodeficiencia, evitando que se presenten complicaciones o secuelas de la enfermedad. Para ello se realizó una búsqueda de artículos científicos en las bases de datos bibliográficas PubMed, Scopus, SciELO y Redalyc sobre la deficiencia selectiva de inmunoglobulina A, utilizando como palabras claves de búsqueda: deficiencia de IgA y deficiencia selectiva de IgA, en combinación con: manifestaciones clínicas, diagnóstico, laboratorio, alergia, autoinmunidad, cáncer e inmunodeficiencias. No se realizó exclusión por año de publicación y se revisaron los artículos encontrados de diferentes tipos de estudios. Finalmente, se analizaron las fuentes primarias con las cuales se realizó la síntesis de los aspectos fundamentales para comprender la deficiencia selectiva de IgA, el abordaje diagnóstico, los principales hallazgos de laboratorio y el manejo médico interdisciplinario.
Deficiencia selectiva de inmunoglobulina A
La Sociedad Europea para las Inmunodeficiencias (ESID) define la deficiencia selectiva de IgA (base de datos OMIM #137100 - disponible en https://www.omim.org/en- try/137100) como una inmunodeficiencia primaria, caracterizada por infecciones recurrentes, principalmente de los tractos gastrointestinal y respiratorio, en asociación con una mayor incidencia de manifestaciones alérgicas y autoinmunes en individuos mayores de cuatro años, con niveles de IgA sérica menores de 7 mg/dL y con niveles normales de IgG e IgM, y en quienes se hayan descartado defectos relacionados con los linfocitos T u otras causas de hipogammaglobulinemia 15.
Aún es controvertido el punto de corte que define la concentración patológica de IgA en suero, puesto que el valor actual es el valor sérico mínimo que puede detectarse en la mayoría de los laboratorios. Adicionalmente, aunque la deficiencia selectiva puede manifestarse antes de los cuatro años, se utiliza esta edad mínima para hacer el diagnóstico definitivo, con el objeto de evitar un diagnóstico prematuro en niños menores, en los cuales la deficiencia selectiva de IgA podría ser transitoria, por un desarrollo más lento de las células productoras de IgA en la infancia 16,17.
Manifestaciones clínicas
La deficiencia selectiva de IgA es una enfermedad clínicamente heterogénea y debido a que muchos casos se han detectado entre individuos sanos que acuden a donación de sangre, se ha considerado que la mayoría de ellos son asintomáticos, llevando probablemente a un subregistro de los casos sintomáticos, quienes normalmente no acuden a donar sangre 1. En general, estos pacientes desarrollan con mayor facilidad infecciones en las vías respiratorias y en el intestino, así como enfermedades alérgicas y autoinmunes 18. Los principales hallazgos clínicos y la frecuencia de las principales manifestaciones clínicas encontradas en pacientes con DSIgA se muestran en los cuadros 1 y 2.
Cuadro 1 Hallazgos clínicos asociados con deficiencia selectiva de IgA
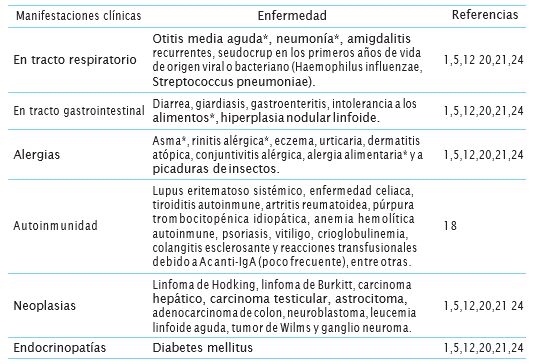
El * indica los hallazgos más frecuentes.
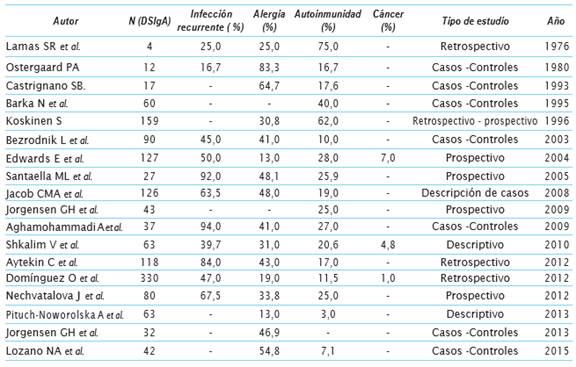
Diagnóstico diferencial
Inicialmente, deben considerarse para evaluación de deficiencia selectiva de IgA los siguientes pacientes: niños con otitis media recurrente, sinusitis o neumonía; adultos con sinusitis crónica e infecciones pulmonares recurrentes; adultos con enfermedad celiaca, infección gastrointestinal por Giardia lamblia y/o fenómenos autoinmunes inexplicables y recurrentes; pacientes con antecedentes familiares de inmunodeficiencia común variable; individuos con antecedentes de reacción anafiláctica a productos sanguíneos 1. Adicionalmente, considerando que las alergias son una de las principales manifestaciones clínicas, planteamos la necesidad de sospechar deficiencia selectiva de IgA en pacientes con alergias múltiples y de difícil manejo. En la consulta se sugiere indagar dentro de los antecedentes familiares por casos de otros errores innatos de la inmunidad o de autoinmunidad. Sin embargo, para confirmar el diagnóstico fenotípico y descartar una inmunodeficiencia común variable debe realizarse dosificación de inmunoglobulinas séricas, preferiblemente por nefelometría, pues tiene mayor sensibilidad 19. Debido a que varios estudios han reportado que entre el 5-20 % de los casos de deficiencia selectiva de IgA se asocian a deficiencia de una o varias subclases de IgG, se deberían cuantificar estas inmunoglobulinas 20 - 22.
Por otra parte, debe evaluarse la determinación de la respuesta a la vacunación, ya sea a antígenos proteicos (difteria, tétanos) como polisacáridos (neumococo), puesto que cualquier anormalidad en este contexto excluiría el diagnóstico de deficiencia selectiva de IgA 15. De igual manera, el estudio de las subpoblaciones de linfocitos T en sangre periférica está indicado en estos pacientes, ya que defectos en ellos excluyen el diagnóstico de deficiencia selectiva de IgA 15. Es importante realizar los exámenes paraclínicos para excluir causas secundarias de hipogamaglobulinemia e inmunodeficiencias asociadas y, adicionalmente, determinar si el paciente con sospecha de deficiencia selectiva de IgA cumple los criterios de diagnóstico establecidos internacionalmente 15. Por lo tanto, proponemos el siguiente algoritmo clínico o diagrama orientador que servirá como guía para tomar decisiones diagnósticas y terapéuticas (figuras 1 y 2).
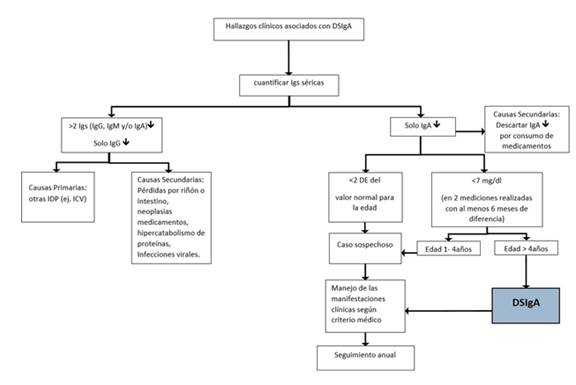
Igs (inmunoglobulinas), (↓) disminución, IDP: inmunodeficiencia primaria, ICV: inmunodeficiencia común variable, DE: desviación estándar.
Figura 1. Algoritmo clínico para el abordaje inicial de un paciente con sospecha de DSIgA. Inicialmente proceder a cuantificar inmunoglobulinas (Igs) séricas. Niveles de IgA sérica <7 mg/dL en al menos dos mediciones, realizadas en un lapso mayor a seis meses, con valores séricos de IgG e IgM dentro de los rangos de normalidad establecidos para la edad, en un niño mayor a cuatro años apoyan el diagnóstico. De otra manera, se considera caso sospechoso. En cualquier circunstancia, descartar otras causas de hypogammaglobulinemia y realizar seguimiento anual de las Igs séricas si persiste la clínica sugestiva.
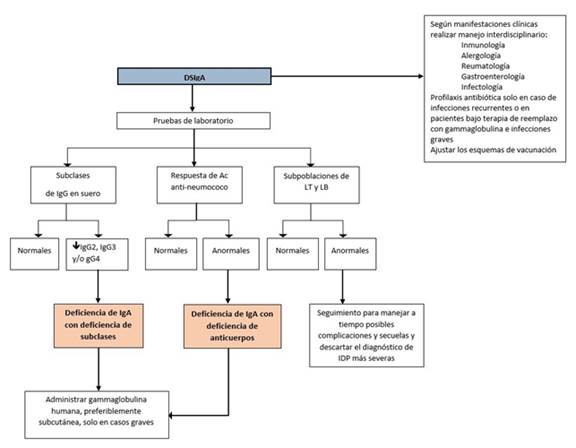
DSIgA: deficiencia seleciva de IgA , ICV: Inmunodeficiencia común variable, Ac: Anticuerpos, LT: Linfocitos T, LB: Linfocitos B, IDP: inmunodeficiencia primaria.
Figura 2 Algoritmo clínico para el diagnóstico definitivo y manejo de los pacientes con DSIgA. Es importante realizar un manejo interdisciplinario de las manifestaciones clínicas del paciente para evitar com- plicaciones o secuelas posteriores. Se debe definir si se realiza profilaxis antibiótica según los criterios ya establecidos y ajustar los esquemas de vacunación. Al momento del diagnóstico se deben realizar idealmente los exámenes enlistados para orientar algunas conductas.
Manejo clínico
Los individuos con diagnóstico de deficiencia selectiva de IgA asintomáticos no necesitan un tratamiento específico, solo manejo expectante 23 y se recomienda una evaluación por inmunología cada cuatro a seis meses 24. En cuanto a los pacientes sintomáticos, estos deben recibir manejo interdisciplinario por alergología, reumatología, gastroenterología e infectología, así como seguimiento al menos una vez al año de los hallazgos clínicos e inmunológicos para controlar la aparición de nuevas manifestaciones asociadas y la evolución a inmunodeficiencia común variable.
Respecto a la vacunación, los pacientes con deficiencia selectiva de IgA responden adecuadamente a todas las vacunas y, particularmente, se recomienda la vacuna contra Streptococcus pneumoniae y Haemophilus influenzae tipo B. Con respecto a las vacunas vivas, la vacuna oral contra la polio está contraindicada debido al riesgo de- mostrado de excreción prolongada del virus. Además, se recomienda no administrar BCG y la vacuna contra la fiebre amarilla. Las otras vacunas vivas parecen ser segu- ras, pero deben ser administradas con precaución 25. Actualmente, no es posible el reemplazo de la IgA en pacientes con deficiencia selectiva de IgA, aunque sería interesante determinar si esta terapia de reemplazo (ya sea por vía oral o tópica) podría ser beneficiosa, considerando la corta vida media de esta inmunoglobulina y la tendencia de los pacientes de desarrollar anticuerpos anti-IgA.
Conclusión
Para un adecuado diagnóstico de la deficiencia selectiva de IgA, se debe tener en cuenta que los pacientes se caracterizan por infecciones recurrentes, de los tractos gastrointestinal y respiratorio, en asociación con manifestaciones alérgicas y autoinmunes en individuos mayores de cuatro años, con niveles de IgA sérica menores de 7 mg/dL y con niveles normales de IgG e IgM, y en quienes se hayan descartado defectos relacionados con los linfocitos T u otras causas de hipogammaglobulinemia. Con respecto al manejo clínico, se deben ajustar los esquemas de vacunación e implementar profilaxis antibiótica en las infecciones graves y recurrentes. Adicionalmente, para mejorar el pronóstico, se debe realizar una atención del paciente por un equipo médico interdisciplinario y un seguimiento continuo por un prolongado periodo de tiempo.

