











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introducción
La neurofibromatosis (NF) es un desorden genético de las células derivadas de la cresta neural, afecta el crecimiento de los tejidos neurales de piel y del sistema nervioso central. Se conocen tres tipos: NF1, NF2 y Schwannomatosis 1.
La NF1 ocurre en 1 de cada 3 000 a 4 000 personas 2, se presenta desde la infancia o en transcurso de la vida, con lesiones que persisten hasta la vida adulta. En pacientes con NF se ha encontrado una disminución en la esperanza de vida de hasta 15 años comparado con la población general, por su asociación con neoplasias malignas y enfermedad vascular 3.
La neurofibromatosis segmentaria (NFS) se describió inicialmente como neurofibromatosis sectorial por Crowe et al. en 1956 y posteriormente en 1977 se denominó como Neurofibromatosis segmentaria por Miller y Sparkes 4. A su vez, Riccardi subdivide la NF en ocho subtipos, incluyendo la NFS como NF tipo V, además describió los hallazgos clínicos como: manchas cafés con leche y/o neurofibromas en un segmento unilateral del cuerpo, sin cruzar la línea media, en pacientes sin historia familiar ni compromiso sistémico 5.
En 1987 Rothen subdivide la NFS en cuatro subtipos: segmentaria verdadera, localizada con compromiso profundo, hereditaria y bilateral 6.
Por la gran variedad de manifestaciones y variantes atípicas de NF, Weiss y colaboradores proponen una nueva clasificación: NF1 clásica donde hay una deleción completa del gen NF1 y formas alternas de NF1 (con características incompletas atípicas), en donde se incluye la NFS 7.
La prevalencia de NFS estimada es tan baja como 1 por cada 40 000 personas en la población general, aunque también se ha considerado subdiagnosticada por su curso indoloro y asintomático 8.
Se plantea que la NFS no tiene componente hereditario y se debe a una mutación postcigótica en el gen NF1, y que el momento cronológico en que sucede la mutación se asocia con el grado de compromiso de la enfermedad, siendo las mutaciones tardías en la embriogénesis las que dan como resultado las formas localizadas 9.
Se ha descrito la NFS como una entidad benigna sin afección sistémica, aun así, puede haber compromiso de órganos (especialmente en pacientes con neurofibromas plexiformes) e incluso presentaciones familiares, debidas a que la mutación gonadal se manifiesta en descendencia como enfermedad generalizada 10.
Presentación del caso
Paciente de sexo femenino de 63 años, quien cursa con dermatosis de ocho años de evolución que afecta el tronco posterior de manera unilateral a nivel de los dermatomas T10-T11, caracterizada por múltiples neoformaciones exofíticas de 2 a 3 mm de diámetro, en forma de domo, del color de la piel y de consistencia blanda, depresibles a la palpación (Figura 1). Refiere que previamente se realizó electrodesecación de algunas lesiones, con recurrencia de las mismas, pero han permanecido asintomáticas por lo que no solicita tratamiento.
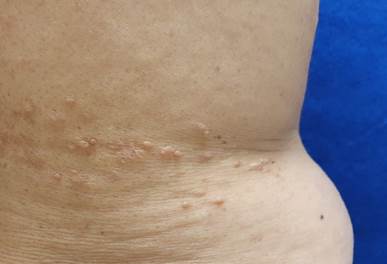
Figura 1 Dermatosis localizada a el tronco afecta espalda a nivel de los dermatomas T11-T12, consistente en neoformaciones de aspecto papular milimétricas del color de la piel, con bordes definidos, a la palpación de consistencia blanda.
Se tomó biopsia diagnóstica de una de las lesiones y en la histología se reportó una epidermis anfractuosa con pigmentación de la capa basal, en la dermis reticular se evidenciaron fibras de colágena hialinizada con algunas células de aspecto fusiforme, dispersas; en la dermis reticular media a profunda se aprecian fibras de colágena engrosadas, entremezcladas con algunas células fusiformes. (Figura 2-3 y 4).
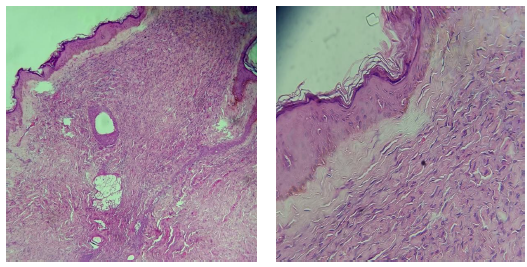
Figura 2-3 HyE 4- 10x, El estudio histológico mostró una neoformación no encapsulada, localizada en la dermis reticular, compuesta por fibras de colágena hialinizada con células fusiformes entremezcladas con citoplasma claro y núcleo ondulado.
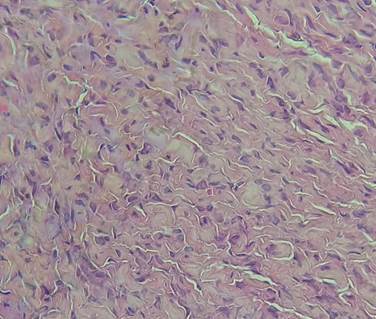
Figura 4 HyE 40x. En este acercamiento se observan fibras de colágena engrosadas entremezcladas con células fusiformes de citoplasma eosinófilo, algunas con núcleo estrellado, otras con núcleos ondulados o en forma de coma.
La paciente tiene antecedente de hidrocefalia, evaluada por neurología quien dictaminó una ventriculomegalia asociada a hipertensión periventricular de posible origen vascular. Sin lesiones sugestivas de NF1 en sistema nervioso central.
Por parte de oftalmología se descartó compromiso ocular, por lo que se estableció el diagnóstico de neurofibromatosis segmentaria. Posterior al diagnóstico se valoró por parte de genética, refiriendo ausencia de afectación en ascendencia, a su vez, se realizó búsqueda de hallazgos clínicos e imagenológicos en sus hijos y nietos, sin encontrar compromiso en el momento de la revisión.
Discusión
La NFS afecta principalmente el sexo femenino, presentando un pico bimodal de incidencia entre los 10 a 30 años y 50 a 70 años. La topografía más afectada es el tronco, como en el caso de nuestra paciente 11.
Su variabilidad fenotípica permite que se clasifique en cuatro categorías 10: 1. Con cambios solo pigmentarios. 2. Únicamente con neurofibromas. 3. Con neurofibromas y cambios pigmentarios. 4. Con neurofibromas plexiformes.
El diagnóstico de la NFS es principalmente clínico, se presenta con las manchas café con leche y/o neurofibromas en un segmento corporal, sin cruzar la línea media, sin compromiso sistémico ni historia familiar de NF. Los hallazgos histológicos de los neurofibromas, son los de un nódulo delimitado en la dermis o tejido celular subcutáneo, formado por células fusiformes con núcleo ondulado, en un estroma rosa pálido, mucinoso o mixoide 11. El diagnóstico diferencial de los neurofibromas debe realizarse con tumores neurales (schwannoma), leiomioma cutáneo y dermatofibroma 12.
Las indicaciones para la exéresis quirúrgica de un neurofibroma incluyen: dolor, compresión de estructuras adyacentes, desfiguración cosmética, deterioro neurológico y rápido crecimiento sugestivo de degeneración maligna.
En contraste con la NF1 y la NF2, el compromiso sistémico en la NFS es poco común, excepto en pacientes con neurofibromas plexiformes 13. En 2016 Cohen reporta once pacientes con NFS que han desarrollado neoplasias, las más frecuentes fueron el melanoma y los tumores malignos de la vaina de nervio periférico (ambos derivan de células de la cresta neural), cáncer de mama, de colon, gástrico, de pulmón y linfoma de Hodgkin. Se estima que el riesgo de estos pacientes de padecer neoplasias es de un 5% 14.
En una de las series más grandes de pacientes con NFS, de manera retrospectiva incluyeron 37 pacientes, siendo 12 pediátricos y 25 adultos, con seguimiento promedio de 21 años. Reportan ningún paciente con descendencia afecta hasta la fecha del estudio, además de evidenciar ausencia de comorbilidades y afección neoplásica en los casos 15.
Se ha reportado mayor penetrancia en los pacientes que presentan neurofibromas plexiformes y mayor afectación familiar si hay nódulos de Lisch. La incidencia de cáncer en la NFS es similar a la reportada en pacientes con NF1 11,16,17.
Conclusión
La NFS es una patología rara con baja incidencia; subdiagnosticada por su curso asintomático. Hasta ahora se han descrito cuatro subtipos y uno de ellos con penetrancia sistémica variable, por lo cual es importante tener en cuenta esta entidad y hacer seguimiento a pesar de que la mayoría de los casos descritos en la literatura tienen un curso benigno. Además, hay reportes de malignidad asociada a la NFS similar a los pacientes con NF1.
Los pacientes con NFS pueden presentar mutaciones gonadales con manifestaciones en su descendencia, por lo que es importante el estudio genético de todos los pacientes a quienes se les diagnostique esta condición. También cabe resaltar que las manifestaciones pueden aparecer en cualquier momento, por lo que los familiares deben tener un seguimiento en búsqueda de lograr detección temprana.
Pese a que el verdadero riesgo de daño sistémico y neoplasia maligna no está establecido, se sugiere un abordaje multidisciplinario de estos pacientes.

