










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkActa Neurológica Colombiana
versão impressa ISSN 0120-8748
Acta Neurol Colomb. vol.26 no.1 Bogotá jan./mar. 2010
Revisión
Valor genómico de la isocitrato-deshidrogenasa (IDH1/2) en el origen y progresión de los gliomas (OncolGroup)
Genomic value of isocitrate-dehydrogenase (IDH1/2) in glioma origin and progression (ONCOLGroup)
Andrés Felipe Cardona, León Darío Ortiz, Ludovic Reveiz, Jorge Miguel Otero,
Silvia Juliana Serrano, Hernán Carranza, Carlos Vargas, Carlos Castro,
Diana Torres, Carmen Balaña
Andrés Felipe Cardona, MD, MSc, PhD. Grupo Oncología Clínica y Traslacional, Instituto de Oncología, Fundación Santa Fe de Bogotá, Bogotá, D. C., Colombia. Fundación para la Investigación Clínica y Molecular Aplicada del Cáncer (FICMAC), Bogotá, D. C., Colombia; investigador asociado, OncolGroup. Investigador asociado, Red Latinoamericana de Neuro-Oncología (RedLANO).
León Darío Ortiz, MD. Investigador asociado, Red Latinoamericana de Neuro-Oncología (RedLANO). 4 Grupo Oncología Médica, Sección Neuro-Oncología, Instituto Oncología, Clínica Las Américas, Medellín, Colombia.
Ludovic Reveiz, MD, MSc. Fundación para la Investigación Clínica y Molecular Aplicada del Cáncer (FICMAC), Bogotá, D. C., Colombia; investigador asociado, OncolGroup. Investigador asociado, Red Latinoamericana de Neuro-Oncología (RedLANO). Instituto de Investigación y Evaluación de Nuevas Tecnologías, Universidad Nacional de Colombia, Bogotá, D. C., Colombia.
Jorge Miguel Otero, MD. Grupo Oncología Clínica y Traslacional, Instituto de Oncología, Fundación Santa Fe de Bogotá, Bogotá, D. C., Colombia. Fundación para la Investigación Clínica y Molecular Aplicada del Cáncer (FICMAC), Bogotá, D. C., Colombia; investigador asociado, OncolGroup. Investigador asociado, Red Latinoamericana de Neuro-Oncología (RedLANO).
Silvia Juliana Serrano, BSc, MSc. Fundación para la Investigación Clínica y Molecular Aplicada del Cáncer (FICMAC), Bogotá, D. C., Colombia; investigador asociado, OncolGroup.
Hernán Carranza, MD. Carlos Vargas, MD. Carlos Castro, MD. Grupo Oncología Clínica y Traslacional, Instituto de Oncología, Fundación Santa Fe de Bogotá, Bogotá, D. C., Colombia. Fundación para la Investigación Clínica y Molecular Aplicada del Cáncer (FICMAC), Bogotá, D. C., Colombia; investigador asociado, OncolGroup. Investigador asociado, Red Latinoamericana de Neuro-Oncología (RedLANO).
Diana Torres, PhD. Instituto de Genética Humana, Pontificia Universidad Javeriana, Bogotá, Colombia.,
Carmen Balaña, MD, PhD. Grupo Oncología Médica, Sección Neuro-Oncología, Instituto Catalán de Oncología, Hospital Germans Trias I Pujol, Barcelona, España.
Correo electrónico: cbalana@iconcologia.es.
Recibido: 20/01/10. Revisado: 15/02/10. Aceptado: 1/03/10.
RESUMEN
Introduccción. En algunos subtipos de migraña se ha demostrado la existencia de hiperexcitabilidad corLas mutaciones heterocigotas del gen que codifica la isocitrato deshidrogenasa (IDH) ocurren con relativa frecuencia en los gliomas; sin embargo, su relevancia durante el desarrollo tumoral es desconocida. Estas alteraciones provocan una pérdida en la afinidad de la enzima por el sustrato, inhibiendo la actividad de la isoforma silvestre de la IDH1 a través de la formación de heterodímeros inactivos. La expresión forzada de la mutación IDH1/2 en cultivos celulares reduce la formación del producto de la enzima, el α-ketoglutarato (α-KG), e incrementa los niveles del factor inducido por la hipoxia tipo 1 (HIF-1α, un elemento de transcripción que facilita el crecimiento tumoral en presencia de bajas concentraciones de oxígeno, hallazgo regulado en parte por el α-KG. La expresión del HIF-1α suele ser mayor entre los gliomas portadores de la mutación IDH, en los que la vía de señalización del HIF está implicada en su progresión. Varios grupos independientes han demostrado el papel que tienen las mutaciones del gen IDH1/2 como marcador pronóstico, especialmente para los pacientes con gliomas de bajo grado y con glioblastomas secundarios que presentan un patrón oligodendroglial. Este conocimiento proporciona una clara oportunidad para mejorar las estrategias diagnósticas y terapéuticas para los pacientes con gliomas, que en la actualidad no se encuentran dirigidas contra alteraciones moleculares específicas. Este artículo presenta una revisión detallada del papel de las mutaciones del gen IDH en la progresión y el mantenimiento de los gliomas, y explora algunas opciones terapéuticas dirigidas contra este entorno.
PALABRAS CLAVES. mutación, isocitrato deshidrogenasa, factor inducible por la hipoxia, glioma.
SUMMARY
Heterozygous mutations in the gene encoding isocitrate dehydrogenase (IDH) occur in gliomas, but their mechanistic role in tumor development is unknown. Tumor-derived IDH mutations impair the enzyme's affinity for its substrate and dominantly inhibit wild-type IDH1 activity through the formation of catalytically inactive heterodimers. Forced expression of mutant IDH1 in cultured cells reduces formation of the enzyme product, α-ketoglutarate (α-KG), and increases the levels of hypoxia-inducible factor subunit 1 (HIF-1α, a transcription factor that facilitates tumor growth when oxygen is low and whose stability is regulated by α-KG. HIF-1α levels were higher in human gliomas harboring an IDH1 mutation than in tumors without a mutation, thus, IDH1/2 contributes to tumor progression in part through induction of the HIF-1 pathway. Numerous independent research groups had demonstrated the role of IDH mutations as a prognostic marker, especially for those patients with low grade gliomas and secondary glioblastomas with oligodendroglial pattern. This knowledge indicates great opportunities to improve diagnostic and therapeutic strategies for gliomas, which are not currently targeted at the specific molecular alterations. This paper presents a detailed review of the role of the IDH gene mutations in progression and manteinance of gliomas, and explores some therapeutic options directed against this environment.
KEY WORDS. mutation, isocitrate dehydrogenase, hypoxia-inducible factor, glioma.
INTRODUCCIÓN
Según la Agencia Internacional para la Investigación del Cáncer, IARC (Organización Mundial de la Salud - OMS), la incidencia estandarizada de los tumores primarios del sistema nervioso central a nivel mundial es de 15,8/100.000 y 17,2/100.000 habitantes para hombres y mujeres, respectivamente (1). Globalmente estas cifras se traducen en 190.000 casos nuevos por año, 22% más que las neoplasias cerebrales diagnosticadas en 1960, lo cual en la actualidad representa el 1,4% de todos los cánceres (2). En general estas lesiones son más prevalentes entre los habitantes de los países desarrollados y están constituidas, principalmente, por los tumores de estirpe glial (1, 3).
Los gliomas representan la tercera causa de muerte por cáncer entre los hombres de edad media, y la cuarta para las mujeres con edad comprendida entre los 15 y 34 años (4). La supervivencia global (SG) de los pacientes con glioblastoma (GB) y la de aquellos con gliomas anaplásicos (GA) es de 8 a 15 meses y de 12 a 28 meses, respectivamente (5); después de la publicación de Stupp y colaboradores, la gran mayoría de los centros con experiencia en neurooncología utilizan como tratamiento estándar de los gliomas de alto grado la temozolamida (TMZ) en concomitancia con radioterapia, seguida de 6 meses o más de tratamiento con este agente alquilante, con lo que se logra una SG de 14,6 meses y una proporción de sujetos vivos a los 2 años del 26% (6). No obstante, más del 50% de los pacientes progresan durante el primer año, después de lo cual suelen tener una SG menor a 20 semanas (6).
Los gliomas de alto grado presentan un genotipo heterogéneo caracterizado por alteraciones que facilitan su proliferación, expansión y resistencia a diversas intervenciones terapéuticas. Un estudio que valoró el espectro mutacional de 220 pacientes con GB determinó que cerca del 3% del genoma tumoral contiene mutaciones somáticas constituidas principalmente por la sustitución de una base (94%), mientras que el resto incluye inserciones, deleciones y duplicaciones del material genético (7). También se ha confirmado una serie de genes con alteraciones conductoras como el P53, PTEN, CDKN2A, RB1, EGFR, NF1, PIK3CA, y PIK3R1 (8); de éstos, los que presentan alteraciones con mayor frecuencia son el CDKN2A (50%); P53, EGFR, y PTEN (30% a 40%); NF1, CDK4, y RB1 (12% a 15%); PIK3CA y PIK3R1 (8% a 10%) (8).
En paralelo, cerca de 143 genes están sobreexpresados en el GB y 16 codifican para proteínas que participan en las vías de comunicación autocrina y paracrina, o se manifiestan en la membrana celular, lo cual los constituye como candidatos perfectos para el diseño de futuras estrategias terapéuticas o diagnósticas (7). La secuenciación seriada de estos genes en una gran serie de pacientes con GB permitió determinar una elevada frecuencia de alteraciones en el gen IDH1 (isocitrate deshidrogenase-1), localizado en el cromosoma 2q33 y encargado de codificar una enzima que cataliza la carboxilación oxidativa del isocitrato a α-ketoglutarato (α-KG), lo que finalmente resulta en la producción de NADPH (nicotinamide adenine dinucleotide phosphate) (7,8).
De las cinco isoenzimas de la IDH codificadas por el genoma humano, al menos tres se encuentran en la mitocondria (IDH2, 3 y 4), mientras que la IDH1 se localiza en el citoplasma y en los peroxisomas (9). Esta proteína forma un homodímero asimétrico que hace parte del control del daño celular oxidativo a través de la generación de NADPH. En los pacientes con GB la alteración constitutiva del gen IDH1 se encuentra en el 12% y corresponde con una mutación puntual dada por el cambio de una guanina por una adenina en la posición 395 del transcrito (G395A) y por la modificación correspondiente de una arginina por una histidina en el residuo aminoacídico 132 de la proteína (R132H) (9). Esta localización en la proteína está conservada en todas las especies y constituye el punto de unión con su sustrato, que forma un espacio de interacción hidrofílico con el α-carboxilato del isocitrato (10).
A continuación se realiza una descripción detallada de las alteraciones genómicas en el IDH1/2 en los pacientes con gliomas y su repercusión potencial en la práctica clínica.
Alteraciones genómicas en el gen de la isocitrato-deshidrogenasa en pacientes con gliomas
Varias observaciones importantes se han hecho sobre los cambios genómicos en el gen IDH1 en los pacientes con GB; preferentemente ocurren en pacientes jóvenes (media de edad 33 años) con mutaciones concomitantes en el P53, están presentes en casi todos los sujetos con GB secundarios, los portadores tienen un mejor pronóstico —lo que se traduce en una mediana de SG de 45,6 meses frente a 13 meses para aquellos con el genotipo silvestre (IDH1 wild-type; P < 0.001)—, y su presencia es independiente de la activación de otros oncogenes como BRAF, KRAS y PIK3CA (8). Recientemente se describió la inhibición potencial de la IDH1 en pacientes con GB utilizando TNF-α y diferentes agentes de quimioterapia (11).
Tres estudios posteriores confirmaron la presencia de la mutación IDH1 (R132) en más del 70% de los GB secundarios, en buena proporción de los gliomas de bajo grado y ocasionalmente en los GB primarios (5%) (12-14). Balss y colaboradores evaluaron dicha alteración en 685 tumores cerebrales, encontrando la mutación somática de IDH1 en 221 muestras correspondientes a sujetos con astrocitomas difusos (68%), oligodendrogliomas (69%), oligoastrocitomas (78%) y glioblastomas secundarios (88%) (12). Con la intención de confirmar la importancia de este perfil mutacional, Bleeker y colaboradores buscaron la alteración puntual en 672 muestras de tumores, incluyendo gliomas, melanomas, tumores del estroma gastrointestinal (GIST), carcinomas uroteliales, colorrectales, ováricos, pancreáticos, prostáticos, tiroideos, pulmonares y mamarios, al igual que en 84 líneas celulares tumorales de diversos linajes (13). El estudio halló la mutación IDH1 (R132) en el 20% de las muestras correspondientes con gliomas de alto grado y logró identificar tres alteraciones adicionales denominadas R132C, R132G y R132L; sin embargo, el rastreo no evidenció la presencia de estas alteraciones en otras neoplasias. En conclusión, los genes IDH1/2 presentan una función que favorece la generación de los gliomas al ejercer la función de protooncogenes (13).
Recientemente Yan y colaboradores publicaron los resultados de la secuenciación de 939 muestras de tumores, en los cuales se documentó la mutación IDH1 (R132) en 161 casos (14). Las alteraciones se distribuyeron de la siguiente forma: R132H (142 tumores), R132C (7 tumores), R132S (4 tumores), R132L (7 tumores), y R132G (1 tumor). No se encontró ninguna de las mutaciones R132 en 21 muestras de astrocitomas pilocíticos en 2 astrocitomas de células gigantes subependimarios, en 30 ependimomas y en 55 meduloblastomas, ni tampoco, en ninguno de los 494 tumores de otras histologías. El análisis de las mutaciones en el gen IDH2, que codifica la única proteína homóloga a IDH1, y que es capaz de utilizar el NADP+ como aceptor de electrones, demostró 9 mutaciones somáticas localizadas en el residuo R172 (R172G en 2 tumores, R172M en 3 tumores y R172K en 4 lesiones). El residuo R172 de IDH2 es una porción análoga al residuo R132 de IDH1 y está capacitado para generar las mismas alteraciones metabólicas (14).
Para determinar las modificaciones funcionales secundarias a las mutaciones de IDH1/2 se midió la capacidad enzimática de sus proteínas afines (reducción de la NADP+ a NADPH) en una línea celular de oligodendrogliomas transfectados con los genes IDH1 e IDH2 mutantes y silvestres; este procedimiento determinó que la inclusión exógena de IDH1 e IDH2 incrementa de forma significativa la producción de NADPH. Para valorar el papel de las mutaciones del gen IDH en la progresión de los gliomas se valoraron las biopsias seriadas de 7 casos, tomadas durante una etapa temprana de la enfermedad y al momento de la progresión, demostrando que las alteraciones IDH1 (R132) en las lesiones de alto grado deriva de un estado tumoral más temprano (14).
De forma complementaria, se evidenció que el 80% de los astrocitomas anaplásicos portadores de las mutaciones en los genes IDH1/2 presentaron mutaciones en P53, pero sólo el 3% tenía alteraciones en el PTEN (phosphatase and tensin homolog deleted on chromosome 10), EGFR (epidermal growth factor receptor), CDKN2A o CDKN2B (14). En contraposición, los gliomas sin mutaciones en IDH1/2 ocasionalmente exhibieron alteraciones en P53 (18%), y con frecuencia, fueron dominantes las variaciones en el PTEN, EGFR, CDKN2A y CDKN2B (74%). La pérdida de 1p y 19q se encontró en el 85% de los tumores de estirpe oligodendroglial con mutaciones de IDH1/2 pero en ninguna de las lesiones con patrón silvestre (P < 0,001), y entre los gliomas de alto grado los portadores de esta mutación fueron significativamente más jóvenes (mediana de la edad 34; P < 0,001). Las alteraciones de IDH1/2 también condicionaron una mejor SG en los pacientes con GB (31 meses) respecto de aquellos que resultaron ser silvestres (15 meses; P = 0,002) (14). Estos hallazgos fueron similares entre los pacientes con gliomas anaplásicos (SG de 65 meses para los mutados y 20 entre los silvestres; P < 0,001).
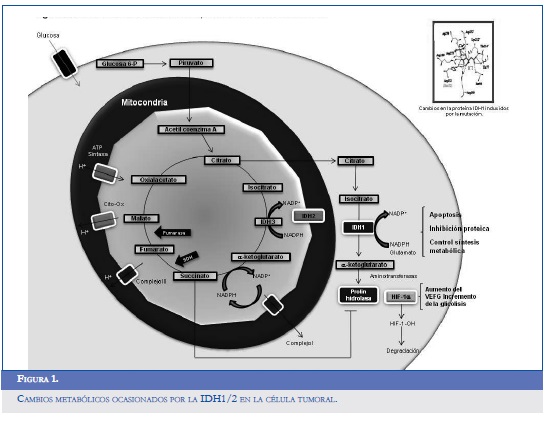
La pérdida de la actividad de IDH1 también se asocia con una reducción de alrededor del 50% en los niveles de α-KG, elemento esencial para el funcionamiento de las polihidroxilasas (PHD), un subgrupo de enzimas encargadas de la hidroxilación y degradación del factor inducido por la hipoxia (hipoxia inducible factor alpha, HIF-1α) (15). Este hallazgo permitió generar una hipótesis sobre la importancia del gen IDH1 como posible estabilizador y modulador de la función del HIF-1α, lo que podría explicar por qué una reducción en la actividad de la α-KG aumenta el estado estable del factor mediador de la angiogénesis tumoral. Dicha presunción se probó tratando células HeLa (línea celular inmortal) con octil-α-KG, un derivado soluble del α-KG que fue capaz de suprimir la producción de HIF-1α alterando los niveles intracelulares de oxígeno y la regulación de la expresión de otros genes implicados en el metabolismo de la glucosa y en el crecimiento tumoral (15, 16). De forma similar, el bloqueo selectivo del gen IDH1 demostró un incremento significativo en la expresión del transportador de glucosa 1 (glucose transporter 1, Glut1), en el factor de crecimiento derivado del endotelio vascular (vascular endothelial growth factor, VEGF), y en la fosfoglicerato quinasa (phosphoglycerate kinase, PGK1) medidos por RT-PCR cuantitativa (Q RT-PCR) (15). En la figura 1 se muestran los cambios metabólicos inducidos por la IDH1 en la célula tumoral.
Para completar esta información, Zhao y colaboradores determinaron si la presencia de las mutaciones de IDH1 se correlaciona con una elevación en los niveles de HIF-1α en 26 pacientes con gliomas; se encontraron 8 tumores portadores de la mutación IDH1 (R132) que tuvieron una expresión de HIF-1α medida por inmunohistoquímica, significativamente superior que los 12 gliomas silvestres (diferencias en la expresión a nivel celular 6,7% ante 3,5%; P < 0,001). Esta relación también fue positiva para la expresión de VEGF (15). Las mutaciones de la IDH1/2 podrían originarse en precursores primitivos de los astrocitos y oligodendrocitos (células tipo A y B) que habitualmente son positivos para nestina y CD133; éstos, a su vez, promueven la proliferación y división por estímulo paracrino en las empalizadas del GB a través de la hipoxia.
Gracias a la estabilización y activación del HIF-1α se produce una hiperplasia endotelial encontrada en el GB que resulta de la translocación y activación de múltiples genes blancos, entre los cuales cabe mencionar el VGFR (vascular endothelial growth factor receptor), las metaloproteinasas de la matriz extracelular (MMP), el inhibidor del factor activador del plasminógeno, el factor transformante α y ß TGF-α/ß, las angiopoietinas, los receptores Tie (Tie-1/2), la endotelina 1 (ET-1), la óxido nítrico sintetasa inducible (IOS), la adrenomedulina y la eritropoyetina (EPO) (16).
Al menos una docena de moléculas diseñadas para inhibir el HIF-1α se hallan actualmente en desarrollo (16), y podrían ser potencialmente útiles para el manejo de pacientes con gliomas que presenten mutaciones en los genes IDH1/2 (17), especialmente después de demostrarse la utilidad de un derivado del α-KG capaz de revertir la producción del factor proangiogénico en cultivos celulares (15). Otras sustancias como el SN-38, que corresponde al metabolito activo del CPT-11 (irinotecan), un medicamento utilizado de forma regular en el tratamiento del GB, ha demostrado su utilidad para inhibir de forma selectiva la proliferación de las células endoteliales tumorales y la expresión de HIF-1α y de VEGF en condiciones de normoxia e hipoxia (18).
Por otra parte, y al igual que otros factores que favorecen la angiogénesis, el HIF-1α también induce la activación de genes comprometidos en la resistencia a múltiples medicamentos (multidrug resistance, MDR); en consecuencia, su control y reducción en cultivos celulares de gliomas transfectados con HIF-1α-siARN (ARN pequeño de interferencia) ha demostrado un incremento substancial de la sensibilidad a varios agentes clásicos de quimioterapia como el etoposido, el cisplatino y la doxorrubicina (19, 20). Un modelo preclínico que combinó la TMZ con un inhibidor selectivo del HIF-1α también demostró un efecto aditivo sobre las células de los gliomas (21).
HIF-1
El HIF es un factor de transcripción encontrado en grandes cantidades en las células de los mamíferos euterios expuestas a bajas tensiones de oxígeno y juega un papel preponderante en la respuesta a la hipoxia; forma un heterodímero constituido por dos subunidades, el componente sensible al oxígeno (alfa) y el HIF expresado de forma constitutiva (subunidad beta; también conocido como ARNT, del inglés aryl hydrocarbon receptor nuclear translocator), que ejerce como colaborador del receptor Ahr (aryl hydrocarbon receptor) (22). El HIF-α se divide en tres proteínas homólogas (HIF1 α, 2, 3); las dos primeras comparten un alto grado de semejanza en sus secuencias, hallazgo que determina una habilidad común para unirse al HIF-ß (23). Poco se sabe del HIF-3α, excepto que se expresa de forma diferencial en diversos tejidos y presenta una variante alternativa conocida como IPAS.
El HIF-1α posee dos dominios encargados de la transactivación, localizados en la porción terminal NH2 (N-TAD, aminoácidos 531-575) y en la porción COOH (C-TAD, aminoácidos 786-826). Durante la normoxia, el gen supresor tumoral Von Hippel-Lindau (pVHL), que hace parte del complejo de ubiquitinación relacionado a las ligasas, hace blanco sobre el HIF-1α favoreciendo su degradación vía proteasoma (24, 25). El dominio para la degradación dependiente de oxígeno (ODDD) que se sobrepone a la fracción N-TAD controla la reducción del HIF-1α; este efecto se ve favorecido por la interacción con la pVHL a través de la hidroxilación de dos residuos de prolina localizados en las posiciones 402 y 564 del ODDD. Otras tres enzimas de la familia PHD (prolyl hydroxylase domain) suprimen el efecto del HIF-1αa nivel postranslacional; todas parecen ser dependientes de la concentración de oxígeno, de la 2-oxoglutarato y del ácido ascórbico. Estudios en in vitro han demostrado el papel de la PHD2 como regulador y estabilizador del HIF-1α, y los PHD 1 y 3 ejercen la función de sensores de la concentración de oxígeno (26). Otras proteínas implicadas en la regulación positiva del HIF-1α son la OS-9 y la ARD1 (ADP ribosylation factor domain protein), y como parte del control negativo el P53, MDM2 y SUMO-1 (small ubiquitin-like modifier-1) (16, 27). La figura 2 resume las vías de activación, interacción y degradación del HIF.
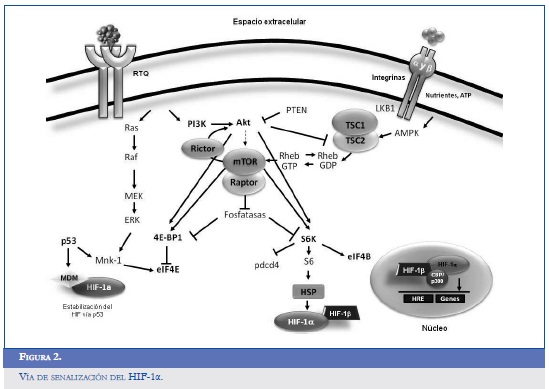
Papel del HIF en la angiogénesis de los gliomas
La activación del EGFR y la pérdida funcional del P53 son hallazgos comunes en los gliomas que presentan sobreexpresión del HIF. La amplificación del EGFR y su mutación más común en los exones 2-7 (EGFRvIII) permite la activación permanente de la vía PI3K/AKT/FRAP/mTOR, que a su vez aumenta los niveles del HIF-1α (28). Los glioblastomas tienen una incidencia de mutaciones en el PTEN que oscila entre el 20% y el 40%, alteraciones relacionadas con un incremento en los niveles del HIF-1α y en la densidad de la microvasculatura tumoral. De forma similar, la pérdida de función del PTEN ocasiona la defosforilación del 3,4,5-trifosfato, que actúa como mensajero para la PI3K, ocasionando la activación constitutiva de las proteínas AKT y MAPK y la consecuente alteración en la regulación del HIF (29). La expresión del P53 silvestre en los gliomas de bajo grado favorece la inhibición del VEGF y del HIF a través del control de la MDM2 vía ubiquitinación (27).
En los gliomas el HIF ejerce la función de un delicado sensor capaz de regular la actividad celular en medio del estrés ocasionado por la hipoxia tisular (16); con el efecto positivo de los bajos niveles de oxígeno se induce la angiogénesis a través de la producción desmedida de VEGF, IGF-II (insulin-like growth factor-II), PiGF (placenta-like growth factor), PDGF-β (platelet-derived growth factor-β), y de las angiopoietinas 1 y 2 (Ang 1/2), que favorecen una rápida proliferación del endotelio y la migración trans-estromal (30). Aparte de su papel en la promoción de la neovasculatura tumoral, el IF-1α también asiste la invasión regulando la expresión de la catepsina-D, la producción de la MMP-2, fibronectina-1, de las queratinas 14, 18 y 19, vimentina, y del factor de motilidad autocrino (31).
Relación entre las células progenitoras de los tumores gliales y el HIF
La proliferación de las células progenitoras de los tumores gliales depende de una interacción dinámica con su microambiente (también llamado “fractona”) (32), en particular, con las estructuras vasculares. Este entorno mantiene un balance irregular entre las señales que favorecen la proliferación de las células tumorales y aquellas antiproliferativas, las cuales representan la clave para controlar la angiogénesis y la homeostasis en los gliomas de alto grado (33). Además, está demostrado que la hipoxia aumenta el flujo cerebral, el consumo encefálico de glucosa y la densidad capilar.
La hipoxia representa uno de los principales indicadores del control de la proliferación de las células progenitoras normales y tumorales; Cipolleschi y colaboradores probaron esta hipótesis al demostrar que las células pluripotenciales de la médula ósea viven en nichos hipóxicos que las protegen de pequeños cambios en la tensión superficial del oxígeno (34). En estados normales los progenitores medulares se desarrollan en espacios donde se presenta una pO2 3% a 5% menor que la de los lugares ricos en células maduras, hallazgo sugerente de que la hipoxia es una condición esencial para inhibir el desarrollo incontrolado y la diferenciación de los precursores (35). Por otra parte, las bajas concentraciones de oxígeno también impiden la transición epitelio-mesenquimal, condición necesaria para promover la migración individual de los primordios neurales, la cual depende de la expresión de proteínas que favorecen la interrelación celular como las integrinas, el receptor para el factor de crecimiento derivado de los hepatocitos (MET), la citocromo oxidasa-2 (cytochrome oxidase-2, COX-2) y las PSA-NCAM (polysialated neural cell adhesion molecules); del último grupo cabe destacar la E-debrina y la E-caderina (36), proteínas parcialmente reguladas por el HIF-1α (37).
Estudios in vitro han encontrado que la comunicación cruzada entre los precursores neurales y las células endoteliales se produce particularmente a través del VEGF, por el factor neutrófico derivado del cerebro (brain-derived neurotrophic factor, BDNF), la neuropilina, algunas semaforinas, y por las moléculas Eph (ephrines) (38). La actividad migratoria de las células progenitoras gliales también depende de la producción de sustancias quimiotácticas y del cambio en su gradiente a nivel del estroma, condición dominada por la producción del HIF, VEGF, PDGF y de sustancias proinflamatorias como el factor CXCL12 (stromal derived factor 1, SDF 1) y su receptor correspondiente, el CXCR4 (39).
En el microambiente tumoral el CXCR4 funciona como un receptor de múltiples quemoquinas que favorecen la supervivencia de las células neoplásicas (40). De igual forma, su activación parece ser dependiente de la hipoxia y de la activación transcripcional del HIF-1α el cual actúa como efector de otras vías que favorecen la inmortalización tumoral, entre otros, las alteraciones del PTEN y en el P53 (41). Así mismo, el HIF está implicado en el reclutamiento de las células TCSC (tissue committed stem cells) que presentan positividad marcada para CXCR4 (CXCR4+) y se encargan de reforzar el proceso de gliogenesis y la producción de nuevos vasos sanguíneos que finalmente degeneran en el crecimiento tumoral y en el rompimiento de la barrera hemato-encefálica causada por las metaloproteinasas (MMP-2 y 9) encontradas en los GB (42).
El nicho tumoral dominado por las células endoteliales y por las TCSC degenera en necrosis, la que a su vez cierra el círculo aumentando la presión interna del tumor y la hipoxia, que resulta en la activación y estabilización del HIF-1α; este evento se traduce en una serie de cambios epigenéticos en los precursores neurales los cuales determinarán finalmente el linaje de la neoplasia glial (43). Estos datos han sido confirmados por observaciones del proceso embrionario de generación del neuroectodermo y de su relación con las células de la cresta neural, zona que da origen a los vasos sanguíneos del sistema nervioso central a partir de cambios en la tensión superficial de oxígeno en la región ventral del tubo neural (44, 45).
Tratamiento antiangiogénico de los gliomas dirigido contra el HIF y otros blancos relacionados
Recientemente Sanson y colaboradores describieron la presencia de la mutación IDH1 (R132) en 132 pacientes con gliomas (100 grado II/77%, 121 grado III/55%, y 183 grado IV/6%), entre quienes también fue frecuente la codeleción del 1p19q y la metilación del promotor del gen MGMT (46); en contraposición, los sujetos sin alteraciones genómicas en la IDH1 presentaron con frecuencia amplificación en el EGFR, la mutación del BRAF y la pérdida del cromosoma 10 (46, 47). La presencia de la mutación IDH1 (R132) favoreció un mejor pronóstico en los sujetos con tumores de bajo grado (SG 150,9 frente a 60,1 meses para los no mutados; P = 0,01), entre aquellos con gliomas de patrón anaplásico (81,1 frente a 19,4 meses; P = 0,001), al igual que en el pequeño subgrupo de sujetos con GB (27,4 frente a 14 meses; P = 0,01), todos con lesiones secundarias de estirpe oligodendrogial. Después de ajustar las variables según el grado tumoral, el estado de la metilación del MGMT, el perfil genómico global y el tratamiento administrado, las mutaciones del IDH1 representaron un marcador pronóstico independiente (HR 0,297, IC95% 0,157-0,564; P = 0,00021). Este estudio fue incapaz de encontrar asociación entre la mutación de la IDH1 y los cambios genómicos en el P53, que usualmente acompañan a los astrocitomas de bajo grado y al GB secundario (46). Por otra parte, la relación entre las alteraciones en la IDH1 y el promotor de la MGMT podría explicarse por la facilidad con la cual se presenta la transición G/A en los tumores metilados, hallazgo que afecta genes críticos como el KRAS, P53 y el MDM2, que finalmente interactúan en las vías de señalización del HIF y con el estrés oxidativo. Estos hallazgos fueron confirmados también por Ichimura y colaboradores, quienes sugirieron la existencia de un subgrupo de tumores gliales que presentan un linaje caracterizado por cambios en la IDH1, p14ARF, MDM2, y en el P53 (48, 49).
Basados en los principios biológicos de una nueva condición patológica podría pensarse en la inhibición directa o indirecta del HIF-1α en los pacientes con gliomas portadores de la mutación IDH1/2, ya sea a través del control de la expresión del mARN de algunas de las proteínas terminales, en los puntos de unión del HIF al ADN, o por la ruptura en la transcripción mediada por este gen (17). La inhibición del HIF-1α en un modelo celular de gliomas (D54MG) diseñado para valorar el sinergismo existente con la TMZ demostró un incremento en la efectividad del alquilante, al evidenciar cambios positivos en la estasis vascular con la posterior disminución del volumen tumoral, además de una regresión en la concentración parcial de oxígeno y en la glucosa, eventos que con frecuencia ocasionan pérdida de la regulación de la homeostasis (50).
Los inhibidores de varias ciclinas dependientes de quinasas han demostrado una reducción en la capacidad migratoria y para la invasión de las células gliomatosas mediadas por el estímulo de la COX-2, VEGF, MMP-2, uPAR y del HIF (51); de forma similar, dos inhibidores de la ILK (integrin-linked kinase, QLT0254 y QLT0267), proteína que hace parte de la vía de señalización del HIF y un HIF-1α-targeted small interfering RNA, han demostrado capacidad para controlar los niveles de expresión de la PKB/Akt y la secreción de VEGF y de HIF, ocasionando un bloqueo en G2-M y la inducción selectiva de la apoptosis (52, 53).
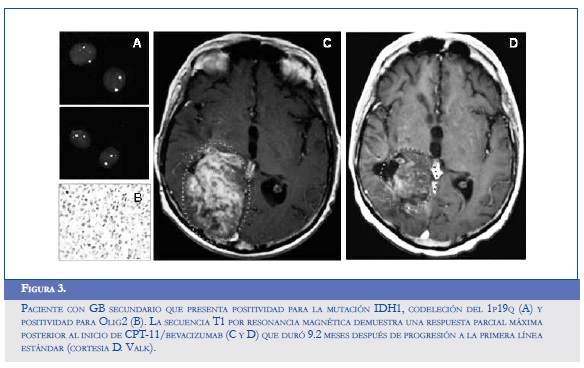
El flavopiridol, un inhibidor de múltiples ciclinas dependientes de quinasas capaz de reducir los niveles de VEGF en el microambiente hipóxico, disminuye en consecuencia la expresión del HIF-1α en las líneas celulares de gliomas U87MG y T98G, que demuestran una elevada capacidad para la invasión y la migración. La reducción de la expresión anormal del HIF también se confirmó después de exponer las células a un inhibidor del proteasoma que usualmente eleva los niveles de esta sustancia proangiogénica (54). Otros modelos celulares han encontrado potencial en el control dual del Ras y del HIF-1α a través del uso de los inhibidores de la farnesil transferasa (trans-farnesylthiosalicylic acid), los cuales también promueven una disminución en la expresión del VEGF y de la Glut-1 (glucose transporter 1), favoreciendo la normalización del proceso de invasión y la glicolisis alterada del GB (55). El radicicol (KF58333) y la geldanamicina, dos antagonistas de la HSP90 (heat shock protein 90), disminuyen la transactivación activa del HIF y promueven su degradación en células gliomatosas independientes de la concentración de oxígeno y de la pVHL (55). Algo similar ocurre al administrar vincristina, paclitaxel, 2-metoxiestradiol, algunos de los inhibidores de la topoisomerasa I y II, y de la PI3K (LY294002) y wortmanina (56). La figura 3 muestra el caso de un paciente con GB secundario que presenta positividad para la mutación IDH1, codeleción del 1p19q (A) y positividad para Olig2 (B). La secuencia T1 por resonancia magnética demuestra una respuesta parcial máxima posterior al inicio de CPT-11/bevacizumab (C y D) que duró 9,2 meses después de progresión a la primera línea estándar.
CONCLUSIONES
Las mutaciones en el gen IDH juegan un papel central en la patogénesis de los gliomas y definen una subpoblación de tumores con particularidades biológicas que se traducen a la clínica (57). El conocimiento recopilado con el estudio del genoma de las neoplasias de estirpe glial representa una oportunidad para mejorar el diagnóstico y las estrategias terapéuticas para los pacientes que desarrollan estas patologías, intervenciones que en la actualidad no están dirigidas contra blancos moleculares específicos relacionados con el control bioenergético de la célula tumoral (58). Los estudios dirigidos a personalizar el tratamiento de los gliomas de bajo y alto grado tendrán que diferenciar las entidades biológicas las cuales poco a poco empiezan a manifestarse en el océano de información traslacional que representa la oncología actual. Estas diferencias se mantienen entre las poblaciones pediátricas y de adultos, sugiriendo que el origen de los gliomas tiene pocos puntos de encuentro en ambos grupos (59).
REFERENCIAS
1. CBTRUS 2000-2004 data. United States population estimates by 5-year age group were obtained from United States census; estimates available at http://www.census.gov. [ Links ]
2. Ries LAG, Melbert D, Krapcho M, Mariotto A, Miller BA, Feuer EJ. et al (eds.). SEER Cancer Statistics Review, 1975-2004, National Cancer Institute. Bethesda, MD, http://seer.cancer.gov/csr/1975_2004/, based on November 2006 SEER data submission, posted to the SEER web site, 2008. [ Links ]
3. Ferlay J, Bray F, Pisani P, Parkin DM. Globocan 2002: Cancer Incidence, Mortality and Prevalence World Wide, Version 2.0. IARC CancerBase No. 5. Lyon, IARCPress, 2004. Limited version available from: URL: http://www_depdb.iarc.fr/globocan2002.htm. [ Links ]
4. Prados MD, Wilson CB. Neoplasms of the central nervous system. In Holland JF, eds. Cancer Medicine. Philadelphia, London: Lea and Febiger 1993: 1080-1119. [ Links ]
5. Gilbert MR. Designing clinical trials for brain tumors: the next generation. Curr Oncol Rep 2007; 9(1):49-54. [ Links ]
5. Stupp R, Mason WP, Van den Bent MJ, Weller M, Fisher B, Taphoorn M et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005; 352: 987-996. [ Links ]
6. Wong ET, Hess KR, Gleason MJ, Jaeckle KA, Kyritsis AP, Prados MD et al. Outcomes and prognostic factors in recurrent glioma patients enrolled onto phase II clinical trials. J Clin Oncol 1999; 17: 2572-2578. [ Links ]
7. Parsons DP, Jones S, Zhang X, Cheng-Ho Lin J, Leary RJ, Angenendt P et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008; 321: 1807-1812. [ Links ]
8. The Cancer Genome Atlas (TCGA) Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008; 455(7216): 1061-1068. [ Links ]
9. Geisbrecht BV, Gould SJ. The human PICD gene encodes a cytoplasmic and peroxisomal NADP(+)-dependent isocitrate dehydrogenase. J Biol Chem 1999; 274(43): 30527-30533. [ Links ]
10. Nekrutenko A, Hillis DM, Patton JC, Bradley RD, Baker RJ. Cytosolic isocitrate dehydrogenase in humans, mice, and voles and phylogenetic analysis of the enzyme family. Mol Biol Evol 1998; 15(12): 1674-1684. [ Links ]
11. Kil IS, Kim SY, Lee SJ, Park JW. Small interfering RNA-mediated silencing of mitochondrial NADP+-dependent isocitrate dehydrogenase enhances the sensitivity of HeLa cells toward tumor necrosis factor-alpha and anticancer drugs. Free Radic Biol Med 2007; 43(8): 1197-1207. [ Links ]
12. Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, Von Deimling A. Acta Neuropathol 2008; 116(6): 597-602. [ Links ]
13. Bleeker FE, Lamba S, Leenstra S, Troost D, Hulsebos T, Vandertop WP, Frattini M et al. IDH1 mutations at residue p.R132 (IDH1(R132)) occur frequently in high-grade gliomas but not in other solid tumors. Hum Mutat 2009; 30(1): 7-11. [ Links ]
14. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009; 360(8): 765-773. [ Links ]
15. Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1α. Science 2009; 324: 261-265. [ Links ]
16. Kaur B, Khwaja FW, Severson EA, Matheny SL, Brat DJ. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro-Oncol 2005; 7(2): 134-153. [ Links ]
17. Semenza GL. Evaluation of HIF-1 inhibitors as anticancer agents. Drug Discov Today 2007; 12(19-20): 853-859. [ Links ]
18. Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev 2007; 21(21): 2683-2710. [ Links ]
19. Kamiyama H, Takano S, Tsuboi K, Matsumura A. Anti-angiogenic effects of SN38 (active metabolite of irinotecan): inhibition of hypoxia-inducible factor 1 alpha (HIF-1alpha)/vascular endothelial growth factor (VEGF) expression of glioma and growth of endothelial cells. J Cancer Res Clin Oncol 2005; 131(4): 205-213. [ Links ]
20. Chen L, Feng P, Li S, Long D, Cheng J, Lu Y, Zhou D. Effect of hypoxia-inducible factor-1alpha silencing on the sensitivity of human brain glioma cells to doxorubicin and etoposide. Neurochem Res 2009; 34(5): 984-990. [ Links ]
21. Li L, Lin X, Shoemaker AR, Albert DH, Fesik SW, Shen Y. Hypoxia-inducible factor-1 inhibition in combination with temozolomide treatment exhibits robust antitumor efficacy in vivo. Clin Cancer Res 2006; 12(15): 4747-4754. [ Links ]
22. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxiainducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. PNAS 1995; 92: 5510-5514. [ Links ]
23. Iwai K, Yamanaka K, Kamura T, Minato N, Conaway RC, Conaway JW, Klausner RD et al. Identification of the von Hippel-lindau tumor-suppressor protein as part of an active E3 ubiquitin ligase complex. PNAS 1999; 96: 12436-12441. [ Links ]
24. Lisztwan J, Imbert G, Wirbelauer C, Gstaiger M, Krek W. The Von Hippel-Lindau tumor suppressor protein is a component of an E3 ubiquitin-protein ligase activity. Gen Develop 1999; 13: 1822-1833. [ Links ]
25. Patiar S, Harris AL. Role of hypoxia-inducible factor-1α as a cancer therapy target. End Rel Cancer 2006; 13: S61-S75. [ Links ]
26. Metzen E, Berchner-Pfannschmidt U, Stengel P, Marxsen JH, Stolze I, Klinger M et al. Intracellular localization of human HIF-1 alpha hydroxylases: implications for oxygen sensing. J Cell Scien 2003; 116: 1319-1326. [ Links ]
27. Ravi R, Mookerjee B, Bhujwalla ZM, Sutter CH, Artemov D, Zeng Q et al. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev 2000; 14: 34-44. [ Links ]
28. Clarke K, Smith K, Gullick WJ, Harris AL. Mutant epidermal growth factor receptor enhances induction of vascular endothelial growth factor by hypoxia and insulin-like growth factor-1 via a PI3 kinase dependent pathway. Br J Cancer 2001; 84: 1322-1329. [ Links ]
29. Sansal I, Sellers WR. The biology and clinical relevance of the PTEN tumor suppressor pathway. J Clin Oncol 2004; 22: 2954-2963. [ Links ]
30. Kelly BD, Hackett SF, Hirota K, Oshima Y, Cai Z, Berg-Dixon S et al. Cell type-specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in nonischemic tissue by a constitutively active form of hypoxia-inducible factor 1. Circ Res 2003; 93: 1074-1081. [ Links ]
31. Krishnamachary B, Berg-Dixon S, Kelly B, Agani F, Feldser D, Ferreira G et al. Regulation of colon carcinoma cell invasion by hypoxia-inducible factor 1. Cancer Res 2003; 63: 1138-1143. [ Links ]
32. Mercier F, Kitasako JT, Hatton GI. Anatomy of the brain neurogenic zones revisited: fractones and the fibroblast/macrophage network. J Comp Neurol 2002; 451: 170-188. [ Links ]
33. He XC, Zhang J, Li L. Cellular and molecular regulation of hematopoietic and intestinal stem cell behavior. Ann N Y Acad Sci 2005; p. 28-38. [ Links ]
34. Cipolleschi MG, Dello Sbarba P, Olivotto M. The role of hypoxia in the maintenance of hematopoietic stem cells. Blood 1993; 7: 2031-2037. [ Links ]
35. Studer L, Csete M, Lee SH, Kabbani N, Walikonis J, Wold B et al. Enhanced proliferation, survival, and dopaminergic differentiation of CNS precursors in lowered oxygen. J Neurosci 2000; 19: 7377-7383. [ Links ]
36. Dirks PB. Glioma migration: clues from the biology of neural progenitor cells and embryonic CNS cell migration. J Neurooncol 2001; 2: 203-212. [ Links ]
37. Diabira S, Morandi X. Gliomagenesis and neural stem cells: Key role of hypoxia and concept of tumor "neo-niche''. Med Hypot 2008; 70: 96-104. [ Links ]
38. Ward NL, Lamanna JC. The neurovascular unit and its growth factors: coordinated response in the vascular and nervous systems. Neurol Res 2004; 26: 870-883. [ Links ]
39. Ehtesham M, Yuan X, Kabos P, Chung NH, Liu G, Akasaki Y et al. Glioma tropic neural stem cells consist of astrocytic precursors and their migratory capacity is mediated by CXCR4. Neoplasia 2004; 3: 287-293. [ Links ]
40. Zhou Y, Larsen PH, Hao C, Yong VW. CXCR4 is a major chemokine receptor on glioma cells and mediates their survival. J Biol Chem 2002; 51: 49481-49487. [ Links ]
41. Schioppa T, Uranchimeg B, Saccani A, Biswas SK, Doni A, Rapisarda A. Regulation of the chemokine receptor CXCR4 by hypoxia. J Exp Med 2003; 9: 1391-1402. [ Links ]
42. Schneider SW, Ludwig T, Tatenhorst L, Braune S, Oberleithner H, Senner V et al. Glioblastoma cells release factors that disrupt blood-brain barrier features. Acta Neuropathol (Berl) 2004; 3: 272-276. [ Links ]
43. Diabira S, Morandi X. Gliomagenesis and neural stem cells: Key role of hypoxia and concept of tumor "neo-niche''. Med Hypot 2008; 70: 96-104. [ Links ]
44. Sanai N, Álvarez-Buylla A, Berger MS. Neural stem cells and the origin of gliomas. N Engl J Med 2005; 8: 811-822. [ Links ]
45. Barami K. Relationship of neural stem cells with their vascular niche: Implications in the malignant progression of gliomas. J Clin Neurosc 2008; 15: 1193-1197. [ Links ]
46. Sanson M, Marie Y, Paris S, Idbaih A, Laffaire J, Ducray F et al. Isocitrate Dehydrogenase 1 Codon 132 Mutation is an important prognostic biomarker in gliomas. J Clin Oncol 2009; 27: 4150-4154. [ Links ]
47. Korshunov A, Meyer J, Capper D, Christians A, Remke M, Witt H et al. Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol 2009; 118(3): 401-405. [ Links ]
48. Ichimura K, Pearson DM, Kocialkowski S, Bäcklund LM, Chan R, Jones DTW et al. IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro-Oncol 2009; 11(4): 341-347. [ Links ]
49. Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol 2009. [Epub ahead of print] [ Links ].
50. Li L, Lin X, Shoemaker AR, Albert DH, Fesik SW, Shen Y. Hypoxia-Inducible Factor-1Inhibition in combination with Temozolomidetreatment exhibits robust antitumor efficacy in vivo. Clin Cancer Res 2006; 12(15): 4747-4754. [ Links ]
51. Ali MA, Reis A, Ding LH, Story MD, Habib AA, Chattopadhyay A et al. SNS-032 prevents hypoxia-mediated glioblastoma cell invasion by inhibiting hypoxia inducible factor-1alpha expression. Int J Oncol 2009; 34(4): 1051-1060. [ Links ]
52. Edwards LA, Woo J, Huxham LA, Verreault M, Dragowska WH, Chiu G et al. Suppression of VEGF secretion and changes in glioblastoma multiforme microenvironment by inhibition of integrin-linked kinase (ILK). Mol Cancer Ther 2008; 7(1): 59-70. [ Links ]
53. Fujiwara S, Nakagawa K, Harada H, Nagato S, Furukawa K, Teraoka M et al. Silencing hypoxia-inducible factor-1alpha inhibits cell migration and invasion under hypoxic environment in malignant gliomas. Int J Oncol 2007; (4): 793-802. [ Links ]
54. Newcomb EW, Ali MA, Schnee T, Lan L, Lukyanov Y, Fowkes M, Miller DC et al. Flavopiridol downregulates hypoxia-mediated hypoxia-inducible factor-1alpha expression in human glioma cells by a proteasome-independent pathway: implications for in vivo therapy. Neuro-Oncol 2005; 7(3): 225-235. [ Links ]
55. Blum R, Jacob-Hirsch J, Amariglio N, Rechavi G, Kloog Y. Ras inhibition in glioblastoma down-regulates hypoxia-inducible factor-1alpha, causing glycolysis shutdown and cell death. Cancer Res 2005; 65(3): 999-1006. [ Links ]
56. Powis G, Kirkpatrick L. Hypoxia inducible factor-1A as a cancer drug target. Mol Cancer Ther 2004; 3(5): 647-654. [ Links ]
57. Yan H, Bigner DD, Velculescu V, Parsons DW. Mutant metabolic enzymes are at the origin of gliomas. Cancer Res 2009; 69(24): 9157-9159. [ Links ]
58. Bleeker FE, Atai NA, Lamba S, Jonker A, Rijkeboer D, Bosch KS, Tigchelaar W et al. The prognostic IDH1 (R132) mutation is associated with reduced NADP (+)-dependent IDH activity in glioblastoma. Acta Neuropathol 2010. [Epub ahead of print] [ Links ].
59. Antonelli M, Buttarelli FR, Arcella A, Nobusawa S, Donofrio V, Oghaki H et al. Prognostic significance of histological grading, p53 status, YKL-40 expression, and IDH1 mutations in pediatric high-grade gliomas. J Neurooncol 2010. [Epub ahead of print] [ Links ].
