










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Neurológica Colombiana
Print version ISSN 0120-8748
Acta Neurol Colomb. vol.26 no.1 Bogotá Jan./Mar. 2010
Caso clínico
Síndrome de médula anclada en el adulto
Tethered cord syndrome in adults
Bárbara Aymeé Hernández Hernández, Yenisset Carrero Texidor,
Fara Luisa Cepero Noriega, Miguel Martínez González
Bárbara Aymeé Hernández Hernández. Doctora en Ciencias. Especialista de segundo grado en Fisiología Normal y Patológica (Neurofisiología Clínica). Investigadora auxiliar. Profesora instructora.
Yenisset Carrero Texidor. Máster en Educación Médica Superior. Especialista de primer grado en Fisiología Normal y Patológica. Profesora asistente.
Doctora Fara Luisa Cepero Noriega. Máster en Urgencias Médicas. Especialista de segundo grado en Neurología. Profesora instructora.
Dr. Miguel Martínez González. Máster en Urgencias Médicas. Especialista de primer grado en Ortopedia y Traumatología. Profesor instructor. Departamento de Neurofisiología Clínica. Complejo Científico Ortopédico Internacional Frank País.
Correo electrónico: bibfp@infomed.sld.cu
Recibido: 14/01/10. Revisado: 15/02/10. Aceptado: 16/03/10.
RESUMEN
Introducción. El síndrome de médula anclada es provocado por una fijación anormal de la médula secundaria a disrafismo espinal. Es una afección poco frecuente, de diagnóstico tardío en el adulto, puede confundirse con otras afecciones. Se caracteriza por manifestaciones neurológicas deficitarias sensitivas y motoras de miembros inferiores, deformidades ortopédicas en los pies y estigmas cutáneos. La realización de estudios electrofisiológicos es de vital importancia en estos casos para demostrar el grado de intensidad de la afectación nerviosa, así como su topografía. Las imágenes de resonancia magnética confirman este diagnóstico.
Objetivos. Mostrar casos con síndrome de médula espinal anclada en edades de aparición infrecuente, así como las alteraciones de los estudios neurofisiológicos en estos casos.
Materiales y Métodos. Presentamos tres casos en los cuales la sintomatología aparece en la adultez temprana y se tiene un diagnóstico presuntivo diferente al de médula anclada. A todos estos casos se les realizó estudio de conducción nerviosa periférica, potenciales evocados somatosensoriales y electromiografía de miembros inferiores, así como estudios imagenológicos.
Resultados: Los estudios electrofisiológicos demostraron afección moderada a severa de las estructuras nerviosas evaluadas y las imágenes de resonancia magnética confirmaron el diagnóstico de médula espinal anclada.
Conclusiones. El síndrome de médula espinal anclada es infrecuente en la edad adulta pero debe sospecharse en pacientes que comiencen con deformidades ortopédicas de los miembros inferiores, de curso progresivo, independientemente de la edad de aparición. Existe afectación de grado moderado en los estudios electrofisiológicos, los cuales contribuyen a detectar el grado de afectación neurológica y su topografía.
PALABRAS CLAVES. síndrome de médula anclada, estudio de conducción nerviosa, potenciales evocados somatosensoriales, electromiografía, imágenes de resonancia magnética.
SUMMARY
Introduction. Tethered cord syndrome is due to pathologic fixation of the spinal cord in the spinal canal. It´s an uncommon disease, it´s diagnosis is very late in adults and can simulate other affections. It presents neurological manifestations, orthopedics deformities in legs and skin manifestation. Neurophysiologycal studies are very important in those cases to demonstrate the intensity of nerve affection and exactly topography. The image studies, in special magnetic resonance image confirm this disease.
Objects. Show cases with tethered cord syndrome in uncommon ages, and abnormalities of neurophysiologycal studies in those cases.
Methods. We show three cases with tethered cord syndrome that appears in young ages and others presumptive diagnosis are planted at the beginning of clinical picture. Nerve conduction study, Somatosensory evoked potentials and Electromiography of lower members and Magnetic resonance Images were done to those patients.
Results. Electrophysiologycal studies have showed moderate to severe abnormalities of the nerve structures and Magnetic resonance Images confirmed tethered cord syndrome.
Conclusions. Tethered cord syndrome is uncommon disease in adult but it should be considered in patients that start with progressive orthopedic deformities of lower members. Electrophysiologycal studies are affected. They are very useful to evaluate the neurological structure involvement in it.
KEY WORDS. tethered cord síndrome, Nerve conduction study, Somatosensory Evoked potentials, Electromiography, Magnetic resonance Image.
INTRODUCCIÓN
El síndrome de médula espinal anclada (MEA) es una enfermedad poco frecuente (aparece en el 5% de la población) y de diagnóstico muy tardío en el adulto; incluye un grupo de condiciones patológicas las cuales provocan que el cono medular se encuentre en un nivel anormalmente bajo y fijo en un estado de relativa inmovilidad (1-5).
Es provocado por una fijación anormal de la médula secundaria a disrafismo espinal (6-10).
Las disrrafias constituyen un conjunto de diferentes anomalías de tejido neuroectodérmico, causadas por la alteración de la disyunción del tubo neural durante la embriogénesis; también se denominan defectos del cierre del tubo neural e incluyen fusión imperfecta, además de las estructuras neurales, de estructuras óseas y mesenquimatosas de la línea media (11, 12). En la MEA está dificultada la elongación normal de la médula en sentido cráneo-caudal durante el crecimiento, ejerciéndose tensión sobre los elementos vasculo-neurales y produciéndose déficit neurológico (11, 12).
Formas clínicas:
— Primaria: en estos casos el filum terminal es corto y engrosado y constituye la causa de todo el proceso patológico.
— Secundaria: a afecciones como: espina bífida abierta, lipoma lumbosacro, lipomielomeningocele, lipomielocele, leptomielolipoma, lipomas del filum terminal, tumores intraespinales, meningocele (1, 9, 10, 13, 14).
Fisiopatopatología
En promedio el cono medular se localiza entre L1 y L2 en el adulto normal, igual que en el niño de tres meses de vida extrauterina, cuando en el proceso de ascenso el cono queda detenido o anclado por malformaciones espinales visibles u ocultas. La movilidad medular y radicular restringida da lugar a compromiso de la circulación sanguínea, produciendo isquemia nerviosa y disfunción del metabolismo oxidativo en las interneuronas. Lo mismo puede ocurrir en los niños operados de disrafias espinales en los primeros días de nacidos, en los cuales las adherencias cicatriciales impiden el ascenso de la médula durante el desarrollo ulterior (8, 13-17).
Cuadro clínico
Esta enfermedad tiene un comienzo insidioso e inespecífico; debe considerarse en el diagnóstico diferencial de las neuropatías periféricas y de los síndromes medulares.
Clínicamente esta afección se caracteriza por:
— Manifestaciones neurológicas deficitarias sensitivas y motoras de miembros inferiores.
— Deformidades ortopédicas en los pies (pie cavo, pie equino).
— Estigmas cutáneos, que pueden aparecer en un tercio de los pacientes con MEA. Entre ellos se encuentran el tumor subcutáneo visible en región lumbosacra, pedículo sésil, hemangioma, fosita cutánea, mechón piloso a nivel de la disrafia (4, 5, 16, 18, 19).
Estas manifestaciones pueden aparecer en el momento del crecimiento acelerado (entre 8-10 años) o en la edad adulta como resultado de un trauma o movimiento de flexión exagerado (3, 6, 20-22).
El diagnóstico se basa en la clínica del paciente y en las imágenes de resonancia magnética nuclear (rMN) (20). El tratamiento de este síndrome es eminentemente quirúrgico (13, 19). Entre los exámenes preoperatorios se encuentran los estudios electrofisiológicos, entre ellos, la electromiografía de aguja (EMG), el estudio de conducción nerviosa motor y sensitivo (ECN) y los potenciales evocados somatosensoriales (PESS). Los mismos evalúan la intensidad de afección neurológica, la topografía, permitiendo elaborar un pronóstico de recuperación (2, 4, 7, 11, 15, 18, 23-25). Además, en la mayoría de los casos se utilizan para monitorizar el acto quirúrgico y evitar una lesión neurológica permanente, que pudiese ocurrir al seccionar raíces nerviosas (1, 9, 21, 24-26).
Nos proponemos mostrar tres casos clínicos con diagnóstico de médula espinal anclada con forma de presentación atípica en los cuales se tenía un diagnóstico presuntivo diferente en un inicio por su infrecuencia, así como mostrar el resultado de la alteración de los estudios neurofisiológicos en estos casos.
PRESENTACIÓN DE CASOS CLÍNICOS
Caso clínico 1
Paciente de 17 años, que hace 7 comienza a presentar deformidad de miembros inferiores, fundamentalmente del izquierdo, lo cual le dificulta la marcha.
Examen físico: pies cavos, principalmente el izquierdo, que también es varo. Dedos en martillo. Atrofia de la musculatura intrínseca del pie bilateralmente, especialmente en el izquierdo.
Reflectividad normal, sensibilidad conservada.
Diagnóstico presuntivo: polineuropatía hereditaria.
Rayos X: raquisquisis del sacro, aumento de la lordosis lumbar, espina bífida S1.
RMN: intensidad de señales y morfología de los cuerpos vertebrales lumbares normal. Espacios intervertebrales conservados. Discos intervertebrales normales. No se muestra protrusión ni extrusión de los discos. No se definen alteraciones de los agujeros de conjunción ni de las articulaciones intervertebrales.
Se comprueba la presencia de médula espinal anclada, fija, su porción inferior alcanza el nivel S1-S2, discurriendo en la porción lumbar por la parte posterior del canal raquídeo.
El canal raquídeo se encuentra aumentado en sus ejes, fundamentalmente anteroposterior (34 mm a la altura de L5), dando la impresión de una megacauda.
No se comprueba la presencia de lipomielomeningocele.
Conclusiones: médula espinal anclada.
Estudios neurofisiológicos
Estudio de conducción nerviosa motora (ECNM)
— Prolongación de la latencia de las respuestas motoras del mediano derecho, velocidad de conducción (vCN) enlentecida en forma moderada.
— Respuesta motora del nervio peroneo profundo derecho con latencias muy prolongadas, velocidad enlentecida y amplitud disminuida. Ausencia de respuesta en el izquierdo (Figura 1).
— Respuesta motora de tibial posterior derecho normal y en el izquierdo latencias muy prolongadas y VCN enlentecida.
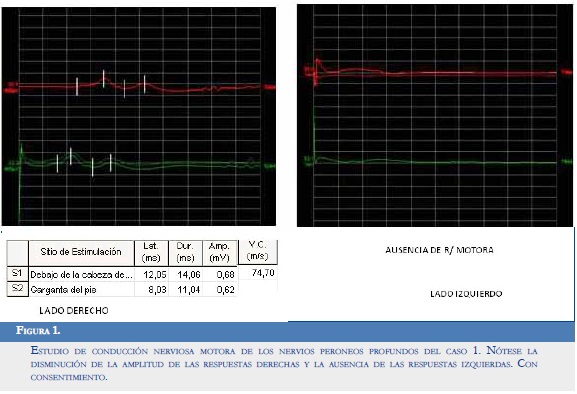
Conclusiones:
— Signos de compromiso mielínico marcado en las fibras motoras del nervio mediano derecho y del tibial posterior izquierdo.
— Signos de compromiso axonomielínico parcial severo de las fibras motoras de ambos nervios peroneos profundos con predominio izquierdo
Estudio de conducción nerviosa sensitiva (ECNS): normal.
PESS de nervio tibial posterior: normal.
EMG de miembros inferiores: potenciales positivos de denervación aislados al reposo.
Potenciales de unidad motora (PUM) mellados y polifásicos de forma difusa. Patrón máximo de contracción aislado en miembro inferior derecho y a oscilaciones simples (OS) en el izquierdo.
Conclusiones: patrón neurógeno moderado en territorio radicular motor L4-S1 bilateral, con ligero predominio izquierdo.
Caso clínico 2
Paciente de 26 años operado de meningocele a los 4 meses de edad. Hace 4 años comienza a presentar dificultad para caminar, deformidad de miembros inferiores, fundamentalmente del izquierdo (pie péndulo izquierdo y varo-equino derecho).
examen físico: cicatriz quirúrgica en la línea media de la región lumbar. Pie péndulo izquierdo y varo-equino derecho. Atrofia de la musculatura intrínseca del pie bilateralmente, especialmente en el izquierdo.
Arreflexia osteotendinosa del miembro inferior izquierdo, sensibilidad conservada.
Diagnóstico presuntivo: deformidad congénita de miembros inferiores.
Rayos X: signos de espondiloartrosis lumbosacra, estrechamiento de los espacios intervertebrales.
RMN: signos de espondiloartrosis lumbosacra, con tendencia a la formación de nódulos intraespinosos, con estrechamiento posterior de casi todos los espacios intervertebrales. Por detrás del cuerpo vertebral de L4-L5 se visualiza una imagen de intensidad heterogénea de 54 x 28 mm que comprime posteriormente el saco dural terminal, impresionando además que el cono medular queda anclado una parte hacia esta imagen, que debe estar en relación con la zona de fibrosis del meningocele operado.
Además, por detrás de L2 se observa también interrupción de la grasa peridural, impresionando atrapamiento de alguna raíz. Todo esto provoca una dilatación de todo el saco dural terminal de forma quística.
Conclusiones:
— Médula espinal o raíces ancladas por fibrosis posquirúrgica por el meningocele operado.
Estudios neurofisiológicos
Estudio de conducción nerviosa motora (ECNM)
— Prolongación de la latencia de las respuestas motoras del mediano derecho, velocidad de conducción (vCN) enlentecida en forma ligera.
— Respuesta motora de ambos nervios peroneos profundos con latencias muy prolongadas, velocidad enlentecida y amplitud muy disminuida con predominio izquierdo.
— Respuesta motora de tibial posterior con latencias prolongadas y vCN enlentecida de forma ligera.
Conclusiones:
— Signos de compromiso mielínico ligero de las fibras motoras del nervio mediano derecho y de ambos tibiales posteriores.
— Signos de compromiso axonomielínico parcial severo de las fibras motoras de ambos nervios peroneos profundos con predominio izquierdo.
Estudio de conducción nerviosa sensitiva (ECNS)
Prolongación ligera de la latencia de la respuesta del sural derecho y vCN enlentecida.
Conclusiones:
— Signos de compromiso mielínico ligero de las fibras sensitivas del nervio sural derecho.
PESS de nervio tibial posterior: ligera prolongación de la latencia de la respuesta P40 derecha y del tiempo de conducción central de ese lado, lo que traduce trastorno de la conducción ligera mediada por fibras mielinizadas gruesas del sistema dorsallemniscal entre la cola de caballo y la corteza del lado derecho (Figura 2).
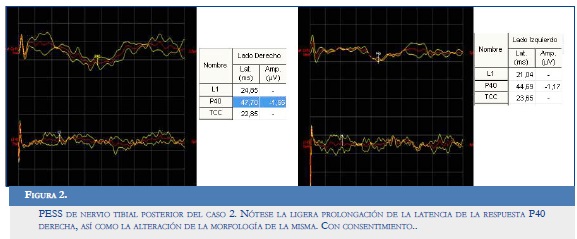
EMG de miembros inferiores: potenciales positivos de denervación dudosos al reposo.
Potenciales de unidad motora (PUM) normales en toda la musculatura explorada. Patrón máximo de contracción aislado en miembro inferior derecho, a OS en gemelo izquierdo. Ausencia de actividad contráctil voluntaria en tibial izquierdo en el miembro izquierdo.
Conclusiones:
— Patrón neurógeno marcado en territorio radicular motor L4-S1 izquierdo y ligero en el derecho.
Caso clínico 3
Paciente femenina de 17 años, con antecedentes de padecer deformidad en los miembros inferiores —sin poder precisar desde cuándo—, lo cual dificulta la marcha.
Examen físico: Pies varos, dedos en martillo.
Hipotonía distal de los miembros inferiores (Figura 3).
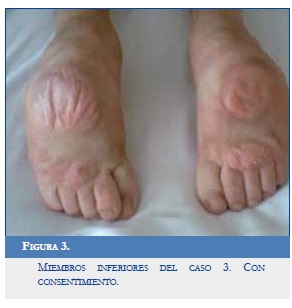
Reflectividad normal, sensibilidad conservada.
Diagnóstico presuntivo: Deformidad en miembros inferiores.
Rayos X: malformación sacra (distrofia del sacro)
RMN: Intensidad normal de señales y morfología de los cuerpos vertebrales lumbares. Espacios intervertebrales conservados. Discos intervertebrales normales. No se muestra protrusión ni extrusión de los discos. No se definen alteraciones de los agujeros de conjunción, ni de las articulaciones intervertebrales.
Se comprueba la presencia de médula espinal anclada, fija, su porción inferior alcanza el nivel S1-S2.
Malformación sacra (distrofia del sacro)
Conclusiones:
— Médula espinal anclada.
Estudios neurofisiológicos
Estudio de conducción nerviosa motora (ECNM)
— respuestas motoras de ambos nervios medianos normales.
— Respuestas motoras de nervios peroneos normales.
— Respuesta motora de ambos nervios tibiales posteriores con amplitudes muy disminuidas y resto de los parámetros normales.
Conclusiones:
— Signos de compromiso axonal parcial severo de las fibras motoras de ambos nervios tibiales posteriores.
Estudio de conducción nerviosa sensitiva (ECNS): Normal.
PESS de nervio tibial posterior: respuestas lumbares normales bilaterales. Ligera prolongación de la latencia de la respuesta P40 bilateral con predominio izquierdo, así como del tiempo de conducción central, lo que traduce trastorno de la conducción ligera mediada por fibras mielinizadas gruesas del sistema dorsal-lemniscal entre la cola de caballo y la corteza bilateral con predominio lado izquierdo.
EMG de miembros inferiores: no se detecta actividad espontánea.
Potenciales de unidad motora (PUM) mellados y polifásicos de forma difusa. Patrón máximo de contracción aislado en ambos miembros inferiores.
Conclusiones:
— Patrón neurógeno moderado en territorio radicular motor L4-S1 bilateral.
DISCUSIÓN
El síndrome de MA es una entidad poco frecuente, pero hay que pensar en ella (1, 9). Nuestro caso número 1, por el cuadro clínico y los estudios electrofisiológicos, fue interpretada como una poli-neuropatía de posible etiología hereditaria. Si no se hubiese completado el estudio con rx y sobre todo con rMN de columna lumbosacra no se hubiese llegado al diagnóstico correcto.
Los casos 2 y 3 fueron interpretados como deformidad ortopédica con afectación de nervios periféricos, a pesar del antecedente referido por el caso 2 de haber padecido y sido operado de meningocele. El estudio de rMN confirmó el diagnóstico de MA.
Desde el punto de vista neurofisiológico, en estos casos existe una afectación predominantemente motora y axonomielínica de los nervios periféricos, fundamentalmente del nervio tibial posterior, que ha sido el afectado de forma más constante y con mayor intensidad.
La afectación observada no se limita al territorio radicular posiblemente comprometido por el anclaje mielorradicular, ya que también existe afectación de los nervios de los miembros superiores.
Al parecer en todos los casos coexisten ambas entidades (MA y neuropatía periférica) o también el daño nervioso periférico pudiese explicarse por una degeneración axonal retrógrada. No hemos encontrado reportes que justifiquen esta asociación, pero al parecer puede ser probable. Aunque en la práctica clínica es una entidad poco frecuente, debe sospecharse siempre la existencia de MA ante un paciente que comienza con deformidades ortopédicas de los pies en edad del crecimiento o en la adultez temprana (1, 9, 10, 19, 26). Tiene gran peso el antecedente de haber padecido o sido operado de alguna malformación espinal, así como la presencia de estigmas cutáneos 5, 14, 24).
Debe realizarse además el estudio imagenológico de rMN, un estudio electrofisiológico que incluya ECNM y S de los nervios de las cuatro extremidades, PESS y EMG para comprobar el estado funcional de las estructuras nerviosas y demostrar la existencia o no de compromiso de los nervios periféricos. Este compromiso, a diferencia del síndrome de MA, no puede corregirse a través del tratamiento quirúrgico sino que necesita además un soporte medicamentoso (2, 7, 11,15, 18, 21, 22, 23, 25).
De no realizarse el diagnóstico oportuno de MA continúa la deformidad ortopédica, ya que la única forma de detenerla es el tratamiento quirúrgico, en el cual, por lo general, se utilizan los estudios electrofisiológicos en el monitoreo transquirúrgico (1, 9, 21, 25, 26).
CONCLUSIONES
— El síndrome de médula espinal anclada puede simular otras afecciones neurológicas.
— Debe sospecharse este síndrome en un paciente que comience con deformidades ortopédicas de los miembros inferiores, de curso progresivo, independientemente de la edad de aparición.
— Existe afectación en grado moderado de los estudios electrofisiológicos, los cuales contribuyen a detectar el grado de afectación neurológica y su topografía.
REFERENCIAS
1. Aboal C, Wilson D, Cibils D, Erman A, Penela M. Cirugía de la médula espinal amarrada con monitoreo neurofisiológico intraoperatorio. Rev Med Urug 2006; 22: 152-56. [ Links ]
2. Aminoff MJ. Electrodiagnosis in Clinical Neurology, 3a ed. New York: Churchill, Livingstone, 1992: 283-327. [ Links ]
3. Aparicio JM. Espina bífida. Protocolos diagnótico terapéuticos de la AEP. Neurología Pediátrica 2008; 129-34. [ Links ]
4. Aparicio M. JM. Espina bífida. Protocolo Nº 18, Neurología, Asociación española de Pediatría. [Consultado el 8 de septiembre de 2007]. Disponible en http://www.aeped.es/protocolos/neurologia/18espina.pdf. [ Links ]
5. Barrero FJ, Salazar S, Ortega MJ, Morales B. Meningitis recurrente como manifestación de un disrafismo espinal en un adulto joven. Rev Neurol, 2002; 35: 825-31. [ Links ]
6. Bassuk AG, Craig D, Jalali A, Mukhopadhyay A, Kim F, Charrow J et al. The genetics of tethered cord síndrome. Am J Med Genet A 2005; 132: 450-3. [ Links ]
7. Binnie CD, Cooper R, Fowler CD, Prior PF. Clinical neurophysiology. EMG, nerve conduction and evoked potentials, 1ª ed. London: Hernemann LTD, 1996: 139-56. [ Links ]
8. Bruce MC. Embriología humana y biología del desarrollo, 3a edición, 2005: 233-275. [ Links ]
9. D´ Agustini Mo, Firreyra ML, Demarchi DH, Emerich JP, Tornesello B, Pennini MG. Monitoreo neurofisiológico intraoperatorio en la cirugía de la médula anclada. Rev Neurocirugía 2006; vIII(2): 48-51. [ Links ]
10. Gutiérrez JJ, Pedroza KG, Cuéllar S. Médula anclada en pacientes pediáricos y adolescentes. Revisión de 16 casos. Rev Med Hosp Gen Mex 2007; 70(2): 62-66. [ Links ]
11. Gutiérrez Rivas E, Jiménez MD, Pardo J, Romer J, editores. Manual de electromiografía clínica. Barcelona: Prus Science; 2000. [ Links ]
12. Herrero AM, Martínez J, Heredero J, Mera J, Masjuán J. Amiotrofia progresiva de una extremidad como síntoma de presentación del síndrome de médula anclada con lipoma espinal. Rev Neurol 2201; 32: 437-40. [ Links ]
13. Kaplan KM, Spivak JM, Bendo JA. Embryology of the spine and associated congenital abnormalities. Spine J. 2005; 5(5): 564-76. [ Links ]
14. Karl FK, Klaus N. Intraoperative monitoring for tethered cord surgery: an Update Neurosurg Focus (series en internet) 2004. [Consultado en abril de 2009]. Disponible en: http://www.medscape.com/viewarticle/470604. [ Links ]
15. Kimura J. Electrodiagnosis in diseases of nerve and muscle: Principles and Practice. Philadelphia; 1983. [ Links ]
16. Ministerio de Salud. Guía clínica. Diagnóstico y tratamiento de disrrafias espinales, 1a edición. Santiago: Minsal; 2005. [ Links ]
17. Otálora BD, Rostion A. Desarrollo embrionario y defectos del cierre del tubo neural. Rev Pediatría electrónica 2007; 4(3). [Consultado en abril de 2009]. Disponible en: http://www.revistamexicandepediatria. [ Links ]
18. Phillips LH, Brown WF, Kimura J. Motor nerve conduction. Cleveland: Cleveland Clinical Foundation, 1995: 21-8. [ Links ]
19. Pilo B, Corral I, Vázquez JM, Rodríguez E. Síndrome de médula anclada en el adulto. Neurología 2006; 21(2): 1-7. [ Links ]
20. Pinto FC, Fontes RB, Leonhardt MC, Amodio DT, Porrof FF, Machado J. Anatomic study of the filum terminale and its correlations with the tethered cord syndrome release in adults. Surg Neurol 2004; 62: 127-33. [ Links ]
21. Quiñones A, Gadkary CA, Gulati M, Von Koch CS, Lyon R, Weinstein PR et al. Neurophysiologycal monitoring for safe surgical tethered cord syndrome release in adults. Sur Neurol 2004; 62: 127-33. [ Links ]
22. Ramírez JM, Alarcón G, Valdevenito G, Cárdenas N, Flandez A. Médula anclada de presentación atípica. A propósito de un caso clínico. Scemess 2005; 1(1): 39-42. [ Links ]
23. Santos CM. El abecé de la electroneuromiografía clínica. La Habana: Ciencia Médicas; 2000. [ Links ]
24. Selden NR, Nixon RR, Skoog SR, Lasheley DB. Minimal tethered cord síndrome associated with thickening of the terminal filum. J. Neurosurg Pediatrics 2006; 105: 214-18. [ Links ]
25. Stalberg E, Falck B. Clinical motor nerve conduction studies. Methods in Clinical Neurophysiology 1993; 4: 61-80. [ Links ]
26. Yamada S. What is the true tethetered cord síndrome? Childs Nerv Syst 2007; 23: 371-75. [ Links ]
