










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkActa Neurológica Colombiana
versão impressa ISSN 0120-8748
Acta Neurol Colomb. vol.26 no.2 Bogotá abr./jun. 2010
Caso clínico
Actualidad en la patología y tratamiento de los gangliogliomas: ¡un tumor no tan benigno!
Update on treatment and pathology of gangliogliomas: !A not so benign tumor!
Fernando González Trujillo, Pedro Penagos González, Camilo Zubieta, Gonzalo Melo Gómez, Alfredo Ernesto Romero Rojas, Gonzalo Guevara, Fabián Neira, Jimy Medina
Fernando González Trujillo, Neurología - profesional observador en Neurooncología. Instituto Nacional de Cancerología, Hospital Simón Bolívar, Médicos Asociados, Clínica Fundadores.
Pedro Penagos González, Coordinador Unidad de Neurocirugía - Oncología. Instituto Nacional de Cancerología, Organización Sanitas, Clínica Reina Sofía.
Camilo Zubieta, Neurocirugía - Oncología. Instituto Nacional de Cancerología, Clínica de Occidente.
Gonzalo Melo Gómez, Neurología - Oncología. Instituto Nacional de Cancerología, Clínica Navarra.
Alfredo Ernesto Romero Rojas, Patología - Oncología. Instituto Nacional de Cancerología, Clínica Palermo.
Gonzalo Guevara, Médico Genetista. Instituto Colombiano de Genética y Oncología Molecular.
Fabián Neira, Médico-Radiología - Intervencionista Oncología. Instituto Nacional de Cancerología, Clínica Fundadores, Clínica Saludcoop.
Jimy Medina, Medicina Interna - Reumatología. Hospital Simón Bolívar.
Correo electrónico: fernando.gonzaleztrujillo@gmail.com
Recibido: 9/09/09. Revisado: 21/09/09. Aceptado: 10/01/10.
RESUMEN
Los gangliogliomas (GG) se definen como tumores mixtos conformados por células de glía y neurona; son considerados como benignos, sin embargo hay reportes de GG que se diferencian por ser tumores de alto grado tipo glioblastoma; este cambio se observa más en el componente glial, pero el componente neuronal puede tenerlo.
La patogénesis de los GG se conoce mejor, parte de esto esta representado por la identificación de cambios en los genes de p53 y la definición de marcadores de diferenciación tumoral como el Ki 67, ambos importantes para el pronóstico y las decisiones terapéuticas.
El tratamiento se basa en una resección quirúrgica amplia considerando la morbilidad del paciente; la quimioterapia y la radioterapia son opciones que han mejorado la sobrevida de los pacientes con GG anaplásicos.
Este manuscrito revisa los cambios en la clasificación histológica y los aportes hechos por la inmunohistoquímica, la genética y las opciones de tratamiento en los GG.
Se presentan dos casos de pacientes, con diferentes edades, para ilustrar el GG benigno y el que se diferencia hacia un tumor de alto grado tipo glioblastoma multiforme, con una detallada revisión patológica.
PALABRAS CLAVES. Patología, sistema nervioso central, neurología.
SUMMARY
Gangliogliomas (GG) are defined as mixed tumors compose from glia and neurons usually with a benign biological behavior. However, there are reports of GG that differentiate toward high grade glioblastoma. This change is observed more frequently in the glial component but neuronal part could be also involved.
Pathophysiology is better understood currently, part of this is represented by the identification of changes in genes at p53 and the definition of tumoral markers like Ki 67, both of them important at the moment of prognosis definition and therapeutic decisions.
Treatment of these tumors implies broad surgical resection due to high morbidity of them. Chemotherapy and radiotherapy are options which improve survival of patients who suffer anaplastic varieties.
This paper review recent changes in histological classification and recent contributions on histology, inmunochemistry, genetics and treatment. In addition we discuss two cases in two different ages and stages showing the usual benign behavior and the transformation to a multiform gliobastoma, along with a detailed pathological review.
KEY WORDS. Pathology, central nervous system, neurology.
Caso clínico
Caso 1. Masculino 29 años. En julio de 2007 consultó el servicio de urgencias por un cuadro clínico de 24 horas con cefalea y vértigo progresivo. Al examen físico tenía bradipsiquia, bradilalia, sin signos meníngeos o focalización motora. Los estudios de TAC e RM cerebral revelaron extensa masa temporoparietal derecha con edema y un gran efecto compresivo. Fue llevado a cirugía tres días después con resección del tumor. La patología mostró un ganglioglioma (OMS ll), el componente ganglionar no presentó atipias, en los elementos gliales había hipercelularidad de tipo astrocitario pero sin evidencia de atipia nuclear, mitosis, ni necrosis.
La evolución fue regular, presentó reacción dérmica severa tipo Steven Johnson a fenitoína, que mejoró. Se realizó una IRM cerebrocontrol en agosto de 2007 que mostró progresión tumoral y se reintervino el 25 de agosto. La revisión patológica de esta muestra fue: imagen histopatológica similar a la anterior pero con llamativa malignización de los elementos gliales. Las áreas ganglioneuronales estaban constituidas por proliferación de células de aspecto ganglionar y neuronal dispuestas en un patrón difuso, sin atipias interpuestas entre un componente glial. Los elementos gliales mostraban el espectro histológico de una lesión de bajo grado (OMS ll) hasta alto grado (OMS lV), en donde se apreciaron los elementos gliales astrocitarios fibrilares dispuestos de forma difusa con atipia nuclear ligera que de forma progresiva adoptaban un mayor pleomorfismo nuclear con incremento de la actividad mitótica, proliferación endotelial vascular glomeruloide y necrosis en empalizada. Con estos hallazgos y la correlación con la clínica e imágenes, se hizo un diagnóstico de tumor glioneuronal mixto maligno de tipo ganglioglioma grado lV con componente glial de alto grado de tipo glioblastoma multiforme, originado sobre la progresión de un astrocitoma fibrilar de bajo grado. Los estudios de inmunohistoquímica mostraron reactividad de los elementos ganglioneuronales para cromogranina, sinaptofisina y PGP 9,5 y para los elementos gliales con PGFA. El índice de proliferación Ki 67 fue de 40% en los núcleos de los astrocitos tumorales.
El análisis citogenético mostró la presencia de dos poblaciones clonales: una hipodiploide y otra hipotetraploide, que deben tener el mismo origen o la segunda evolucionar de la primera por compartir el 6q- como anomalía cromosómica. Desafortunadamente la calidad de las metafases no permitió identificar si existen otras alteraciones cromosómicas. Anexamos imágenes del estudio genético con cariotipo.
La condición del paciente mejoró, el estado funcional -IK- fue del 70%, con síndrome parietal derecho y una hemiparesia izquierda 3/5. Una TAC cerebral de control reveló resección amplia del tumor. Recibió como tratamiento adicional quimioterapia con temozolamida y radioterapia en el lecho tumoral hasta 60 Gy.
La condición general fue estable durante unos meses, luego presentó deterioro neurológico, y una IRM de cerebrocontrol reveló una voluminosa recidiva tumoral. Se decidió no realizar cirugía; el paciente falleció en el año 2008.
Caso 2. Femenino 10 años. En diciembre de 2006 consultó por pérdida súbita de la memoria reciente que duró 24 horas, y mediante una IRM del cerebro se le encontró una lesión tumoral bitalámica. Como antecedente, a los siete años desarrolló pubertad precoz. Se le realizó biopsia por estereotaxia; el reporte de patología fue una lesión grado ll tipo glioneuroma. El tratamiento complementario consistió en quimioterapia de tres ciclos con cisplatino, etopóxido y dexametasona, y tres ciclos complementarios de un esquema "8 en 1". Adicionalmente, recibió radioterapia hasta una dosis de 56 Gy.
Su condición fue estable durante seis meses; en junio de 2007 tuvo convulsiones focales con pánico, parpadeo en ojo derecho, palidez y amnesia. Entró en estado convulsivo generalizado que ameritó UCI; se le aplicó fenitoína y ácido valproico. Fue enviada a la casa con remisión parcial de las convulsiones; se le adicionaron al tratamiento anticonvulsivo tipo clonazepam y clobazam. En julio de 2007 se le practicó una nueva cirugía: se resecó parcialmente la lesión en el tálamo derecho por vía transventricular parieto-occipital y ventriculostomía preventiva. En el segundo reporte de patología los cortes mostraron una neoplasia mixta con componente glial y ganglio neuronal. Las áreas gliales mostraron proliferación difusa de astrocitos fibrilares sin atipia, en donde se reconocían células ganglionares y neuronales sin atipia. No se evidenció mitosis, proliferación endotelial vascular glomeruloide, ni necrosis. Los estudios de inmunohistoquímica mostraron positividad del componente neuronal para sinaptofisina y cromogranina. Los astrocitos fueron positivos para proteína ácida glial fibrilar con un Ki 67 del 1%. El diagnóstico fue el de un ganglioglioma WHO ll.
Se decidió no dar tratamiento adicional. El estado general se deterioró, con persistencia de las convulsiones aún con la adición de otros antiepilépticos (oxcarbamazepina). Falleció en el año 2008.
INTRODUCCIÓN
Los gangliogliomas (GG) son tumores considerados benignos y de lento crecimiento. Se han documentado en todas las edades; en los niños y adultos jóvenes se observa una mayor prevalencia de lesiones con elementos ganglionares mezclados con elementos neuronales, y en los adultos mayores son más frecuentes las lesiones con componentes neuronales mezclados con gliomas de alto grado. Predominan en hombres. Organización Mundial de la Salud (OMS) tuvieron relevancia en el género masculino. Los casos de GG anaplásicos que se han dado en mayores de 40 años tienen una sobrevida total corta (1).
Se ubican en el lóbulo temporal pero emergen en cualquier área del sistema nervioso (2, 3). La localización del tumor en estructuras de la línea media —diencéfalo— se considera de mal pronóstico (2). Hay reportes de GG localizados en el tallo cerebral (4) y en el cordón espinal (5).
El tiempo promedio para que un astrocitoma de bajo grado se diferencie hacia la anaplasia tumoral se ha calculado de 4 a 5 años, con una media de sobrevida total de 6 a 8 años (6).
Clínica
La principal manifestación es la convulsión, que puede ser el único síntoma hasta en el 72% de los casos (1, 2). Las neoplasias bitalámicas tienen como signos predominantes hipertensión endocraneana y alteración cognoscitiva (7). Se ha identificado un tipo de paciente cuya evolución clínica es adversa, si tiene el siguiente perfil (1): sexo masculino, más de 40 años de edad, epilepsia refractaria, resección incompleta del tumor y localización extratemporal del tumor, y por histología, hallazgo de atipias (componente gemistocítico) u otros cambios de anaplasia.
Radiología
El l0% de los GG tienen calcificaciones o pequeñas áreas quísticas. En tomografía (TC) sin contraste el centro del tumor es isodenso o hiperdenso, con el contraste se realzan las lesiones que son sólidas y tienen cambios anaplásicos (2); en secuencias de imagen de resonancia magnética (IRM) T1 son hipointensos, isointensos o hiperintensos al cerebro, y en secuencias T2 y densidad de protones son iso o hiperintensos (2, 8).
Los GG quísticos se ubican en cualquier área del cerebro, pero predominan en la fosa craneal media y el cerebelo y tienen calcificaciones en un 63% más que los sólidos. El 50% de los GG quísticos realzan con contraste; el quiste en IRM Tl es hipointenso y en T2 la señal se incrementa. La presencia de edema peritumoral es inusual (8).
Los GG en IRM no tienen una presentación específica, debe sospecharse GG cuando el tumor tiene las siguientes características (9): lesión sólida localizada en el lóbulo temporal con leve o ningún edema, y un realce homogéneo en secuencias de Tl de RM; lesión quística pequeña o masa mixta quística y sólida con pequeño realce o con un nódulo muy realzado.
Patogénesis
La OMS ha definido los gangliogliomas como tumores benignos grado ll. Con la reciente clasificación de los tumores del sistema nervioso central se expandió en forma generosa el grupo de los tumores mixtos glioneuronales y neuronales, e incluyó una serie de entidades que en la anterior versión no estaban reseñados, como fueron el tumor glioneuronal papilar, el neurocitoma extraventricular y el tumor glioneural, formador de rosetas del cuarto ventrículo; lo anterior a consecuencia del desarrollo y utilización de técnicas en inmunohistoquímica las cuales permiten revelar la diferenciación neuronal de los elementos celulares que anteriormente eran clasificados como de origen glial. La descripción de este tipo de lesiones con características mixtas glioneuronales era limitada debido a la carencia de estudios que pudieran confirmar, sin controversia la presencia de elementos mixtos en un mismo tumor. Ahora gracias a la utilización de nuevos marcadores de inmunohistoquímica, tales como PGP 9.5, NeuN, MAP2, PC_BMP y NRP/B, sumados a la clásica utilización de sinaptofisina, cromogranina, N- CAM (CD56), enolasa neuronal específica (NSE) y los neurofilamento (NF), así como a la disponibilidad de nuevas herramientas diagnósticas, se pueden observar estas características en lesiones otrora consideradas unilinaje.
La clasificación actual reconoce ciertas diferencias entre los GG, que los estadifica desde el benigno grado l hasta los anaplásicos grado lll, con una categoría intermedia grado ll atípico que se caracteriza por un aumento de la celularidad, pleomorfismo nuclear, un componente celular glial muy proliferativo pero que no cumple con los criterios para anaplasia tumoral como la necrosis con patrón en empalisada y un alto recambio mitótico (1).
Entre los GG atípicos y anaplásicos hay una "zona gris indeterminada" donde se ubican los tumores con cambios "histológicos atípicos", como son los GG con componente gemistocítico, marcador para atipia que, según los reportes de casos se asocia con una sobrevidad corta libre de progresión; y en el lado opuesto están los GG que contienen droplets de proteínas cuya presencia determina una sobrevida libre de progresión más larga (1). En este grupo pueden incluirse también los GG con componentes de xantoastrocitoma pleomórfico (1).
La patogénesis de los GG no está clara. Hay casos individuales con historia familiar de neurofibromatosis, síndrome de Peutz Jeghers y síndrome de Turcot (1). Los definían como hamartomas por un posible origen en las células de tejido ectópico de la cresta neural (2). Su origen se asoció con la presencia de tejido nervioso autónomo ectópico soportado por los hallazgos de catecolaminas, tirosina hidroxilasa en algunos GG (3).
En los GG se han determinado cambios citogenéticos, pero ninguno es específico. Con base en éstos, se expuso un origen monoclonal (1, 6), basado en los trabajos que evaluaron la inactivación del cromosoma X por prueba de Humara. Otros, como Platten y cols, reportaron en el gene TSC2 (complejo de la esclerosis tuberosa 2) la expresión de un polimorfismo intrónico en el 15% de los pacientes, cambio que sólo se presentaba en el componente glial del tumor (6). Se propuso que la expresión de este alelo predisponía el desarrollo de gangliomas; no obstante, entre los pacientes con epilepsia que tienen GG, muchos no tienen los estigmas característicos de la esclerosis tuberosa (1, 6). En fin, la funcionalidad de esta mutación no está clara (1).
Son tumores mixtos conformados por células gliales y neuronas, en proporción variable. El componente neuronal se reconoce por un abundante citoplasma que contiene sustancia de Nissl y procesos neuríticos argirófilos, que expresan proteínas como neurofilamento y sinaptofisina, las cuales se miden por inmunohistoquímica (3). El componente glial se distribuye en la corteza de manera irregular, y lo conforman células astrocíticas y oligodendrogliales (2, 3). Hay infiltrados con linfocitos perivasculares, vasos sanguíneos telangiectásicos y un componente fibroblástico con extenso depósito de reticulina. Los GG tienen inmunoreactividad perifocal para CD34 que ayuda en el diagnóstico (1, 2).
Los elementos neuronales tienen un patrón bien diferenciado, la evolución agresiva hacia la anaplasia se hace por el componente glial, que exhibe alta actividad proliferativa, reactividad a la proteína p53, y es la causa de recurrencia del tumor (2, 3). Hay reportes de casos donde la transformación anaplásica afectó ambas células: glial y neuronal (10). La presencia de proteínas de la matrix extracelular tipo laminina y colágeno IV se ha relacionado con la capacidad de los GG de extenderse focalmente hacia las leptomeninges y causar gran inflamación perivascular (6). La pérdida en la expresión de la pl9 se ha relacionado con progresión maligna (1).
Entre los marcadores usados para valorar los grados de malignidad tumoral e índices de pronóstico están la p53 y el Ki-67 (índice de proliferación celular); sólo el Ki-67 aportó información relevante de que los astrocitomas grado ll tienen en promedio un ki-67 de 3%, el cual es bajo con respecto a los astrocitomas de alto grado con un Ki-67 mayor a 12% (11). El glioblastoma que emerge de un tumor benigno como el GG tiene cambios característicos en su biología molecular y genética, como los siguientes: pérdida de heterocigocidad (LOH) 10q 63%, amplificación EGFR (receptor factor de crecimiento epidérmico) 8%, deleción p16 19%, mutación p53 65%, mutación PTEN (gen fosfatasa tensin) 4% (11).
La radioterapia es un antecedente determinante en los cambios anaplásicos por el daño que causa en el DNA (1, 10, 12). Estudios de pacientes con GG que progresaron a glioblastomas recibiendo radioterapia mostraron que la mutación en la p53 ocurrió preirradiación y no se pudo concluir qué fue lo determinante para la anaplasia tumoral, tampoco hubo cambios en el gen EGFR (10).
Este grupo de neoplasias mixtas con elementos malignos neuronales o gliales son infrecuentes y no existe en la actualidad un sistema de clasificación aceptado. Sin embargo, Burger (13) propone una clasificación que de forma simple reúne estas malignizaciones en cuatro grupos:
- Tumores de células ganglionares con componentes gliales malignos (de bajo y alto grado o de bajo grado que progresan a alto grado).
- Tumores de células ganglionares con componentes neuronales malignos.
- Gliomas infiltrantes de alto grado con presencia focal de nódulos de neuropilo positivos para sinaptofisina.
- Tumores de células pequeñas con diferenciación neuroblástica focal evidenciada por inmunohistoquímica o microscopia electrónica.
El diagnóstico diferencial de los GG son los tumores de bajo y alto grado, como los astrocitomas difusos, oligodendrogliomas, tumor neuroepitelial disembrioblástico (DNET), astrocitomas pilocíticos y xantoastrocitoma pleomórfico (1).
Tratamiento
La mejor opción es la resección quirúrgica, logrando índices de sobrevida de 7 a 17 años. Cuando se tiene un paciente con tumor benigno y gran morbilidad quirúrgica, la realización de una resección amplia es controvertida, considerando que estas neoplasias son de lento crecimiento. Los estudios evidencian que con una resección amplia o total del tumor se logra mejorar la sobrevida a 5 años hasta en un 80% o más, y que con una resección parcial se corre riesgo de recurrencia de l.4 y el riesgo de muerte se incrementa hasta 5 veces (2, 3, 14). En los pacientes con tumor residual y factores de riesgo para una transformación anaplásica se indica nueva cirugía en forma temprana (1).
La radioterapia se adiciona si hay resección incompleta, recurrencia o cambios anaplásicos en el tumor (2). Cuando el paciente presenta tumor con bajo grado histológico y una resección quirúrgica casi completa, se acepta como opción de manejo sólo observar, hasta evidenciarse progresión tumoral, o realizar radioterapia inmediata. Los grupos RTOG (Radiation Therapy Oncology Group) y la Organización Europea de Investigación y Tratamiento del Cáncer (EORTC) proponen para los tumores de bajo grado que requieren radioterapia una dosis estándar hasta 54 Gy 30 fracciones, y destacan por sus trabajos que la radioterapia temprana sí mejoró la sobrevida libre de progresión pero no cambió la sobrevida total (14). En los tumores con progresión anaplásica el BTCG (Brain Tumor Cooperative Group) propone ofrecer radioterapia de 50 a 60 Gy en dosis de 1,7 a 2 Gy/día, en 5 a 6 semanas. Según las condiciones funcionales del paciente, hay esquemas de radioterapia en concomitancia con quimioterapia (temozolamida) (14).
La quimioterapia en los tumores de bajo grado es controvertida, pero varios estudios demuestran que controla la progresión de los tumores gliales de bajo grado; así, en casos especiales es una alternativa cuando la radioterapia se contraindica por sus efectos secundarios. Los esquemas más usados por los grupos son: vincristina-carboplatino (tasa de respuesta objetiva desde 33% a 43%, tasa de respuesta total entre 74% y 94%, sobrevida libre de progresión 78% a 3 años, cisplatino- etopóxido (tasa de respuesta total 71%, tasa de respuesta objetiva 100%, sobrevida libre de progresión 78% y sobrevida total 100% a 3 años, esquemas de 6 agentes con procarbazina, carboplatino, vincristina, etopóxido, cisplatino, ciclofosfamida (rata de respuesta objetiva 61%, tasa de respuesta total 88%, sobrevida libre de progresión 34%, sobrevida total 61% a 5 años) (15).
DISCUSIÓN
En el caso encontramos que se cumplieron algunos de los factores de riesgo implicados con un mal pronóstico, como son el género masculino, una resección parcial y las atipias, en el reporte de patología. Los índices de anaplasia, como el Ki67 de 40%, superó los promedios que se han reportado para tumores tipo astrocitomas anaplásicos de 12%, y con diferenciación anaplásica hacia un glioblastoma se evidenció que la sobrevida para los pacientes con estos tumores es corta y la respuesta a los tratamientos complementarios como quimioterapia y radioterapia fue pésima.
Desde el punto de vista citogenético, la deleción del brazo largo del cromosoma 6 no es específica de tumores del sistema nervioso, pues se observa con alguna frecuencia en otras neoplasias como leucemias, linfomas y tumores sólidos. Sería de gran importancia que los estudios citogenéticos fueran de rutina en todo tumor del sistema nervioso central.
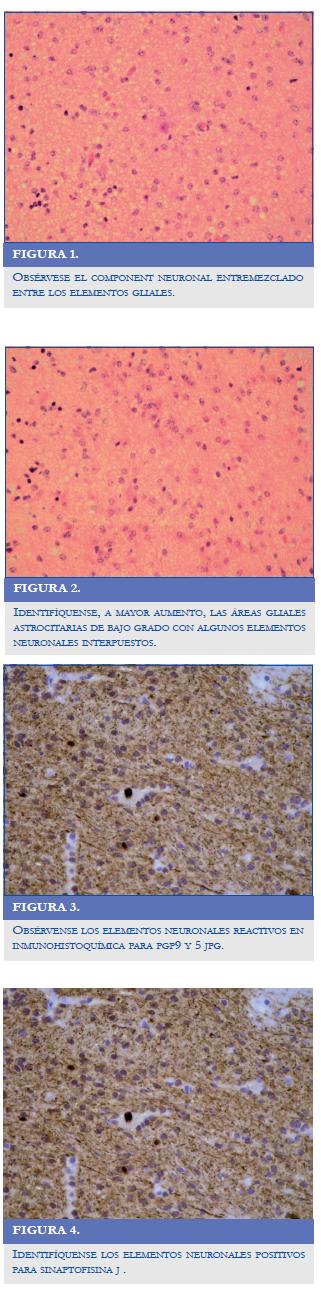
Los gangliogliomas bitalámicos reportados por los grupos de trabajo tienen una evolución muy desfavorable; estas neoplasias representan un grupo tumoral con características clínicas y radiológicas diferentes a los tumores unilaterales, la cirugía se reduce a la realización de una biopsia con propósito diagnóstico, el pronóstico es malo, y sólo el 7,6% de los pacientes sobrevive más de doce meses (7). En el caso, se evidenció cómo un tumor benigno grado ll evolucionó en forma agresiva con mala respuesta a las modalidades terapéuticas ofrecidas, como fueron la quimioterapia y la radioterapia; en los hallazgos de patología no se reportaron cambios sugestivos de anaplasia que explicaran la evolución clínica (Figuras 1-4). Los marcadores de pronóstico y para anaplasia tumoral reportados, como el Ki-67 de 1%, tampoco fue concordante con lo que reporta la literatura.
Hasta la fecha estas lesiones mixtas glioneuronales permanecen como problema en su clasificación pronóstica debido a lo poco frecuente de su presentación, lo cual ha llevado a que muchos aspectos de su comportamiento sean poco claros. Sin embargo, algunos reportes han informado que la expresión de elementos neuronales sólo tiene importancia diagnóstica, sin cambios aparentes en el pronóstico, hipótesis que ha sido debatida ampliamente por otros informes que enuncian un comportamiento más agresivo en las lesiones con componentes neuronales malignos en un aspecto de bajo grado (neurocítico) y de alto grado (neuroblástico).
Los estudios de genética y biología molecular en GG reportan que los cambios más frecuentes y con incidencia en la biología tumoral se dan en la expresión de la proteína p53 y los índices de proliferación celular como el KI-67; en los dos casos revisados no fue posible valorar la p53 por motivo de infraestructura, pero los reportes hasta la fecha no son concluyentes en establecer que la p53 en los GG determine la diferenciación hacia la anaplasia tumoral o sea un factor pronóstico positivo de respuesta a los tratamientos complementarios como la quimioterapia o la radioterapia, pues actualmente la genética y la biología molecular trabajan en la identificación de moléculas o receptores que se expresen en la célula tumoral y sirvan como herramienta terapéutica o para la selección de pacientes que pueden beneficiarse con los esquemas complementarios de tratamiento a la cirugía.
Conclusión
Los GC son un grupo de neoplasias extraordinariamente heterogéneas, con un espectro de presentación muy amplio que abarca grados variables de diferenciación multilinaje, característica reconocida tan solo en años recientes gracias a la utilización generalizada de marcadores de inmunohistoquímica; a pesar de esto, el entendimiento de las características biológicas de los tumores con elementos glioneuronales mixtos es muy pobre, por lo cual se requiere de mayores estudios que aclaren los interrogantes de su génesis, diagnóstico y comportamiento.
REFERENCIAS
1. WOODRUFF WW. Intracranial neoplasms and tumorlike conditions. Fundamentals of neuroimaging. Philadelphia. Saunders Company; 1993: 71-119. [ Links ]
2. MAJORES M, VON LEHE M, FASSUNKE J, SCHRAMM J, BECKER AJ, SIMON M. Tumor recurrence and malignant progression of gangliomas. Cancer 2008; 113: 3355-3363. [ Links ]
3. HAKIM R, LOEFFLER JS, DOUGLAS CA, BLACK PM. Gangliomas in adults. Cancer 1997; 79: 127-131. [ Links ]
4. TRUONG MT. Current role of radiation therapy in the management of brain tumors. Hematol Oncol Clin N Am 2006; 20: 431-453. [ Links ]
5. BURGER PC, SCHEITHAUER BW, VOGEL FS. Neuronal, Glioneuronal, and neurocytic tumors. Surgical Pathology of the Nervous System and its coverings. Boston. Churchill Livingstone; 2002: 290-292. [ Links ]
6. LAGARES A, GÓMEZ P, LOBATO RD, RIOY JR, RAMOS A. Ganglioglioma of the brainstem: report of the three cases and review of the literature. Surg Neurol 2001; 56: 315-322. [ Links ]
7. KARREMANN M, PIETSCH T, JANSSEN G, KRAMM C, WOLFF J. Gangliogliomas. J Neurooncology 2009; 92: 157-163. [ Links ]
8. ZHANG D, HENNING TD, ZOU LG, HU LB, WEN L, FENG XY. Intracranial gangliogliomas: clinicopathological and MRi findings in 16 patients. Clin Radiol 2008; 63: 80-91. [ Links ]
9. PERILONGO G. Considerations on the role of chemotherapy and modern radiotherapy in the treatment of childhood low grade gliomas. Journal of Neuro Oncology 2005; 75: 301-307. [ Links ]
10. HIROSE T, SCHEITHAUER BW, LOPES MB, GERBER HA, ALTERMAT HJ, VANDERBERG SR. Ganglioma An ultrastructural and inmunohistoquimical study. Cancer 1997; 79: 989-1003. [ Links ]
11. HAMBURGER C, BUTTNER A, WEIS S. Gangliogliomas of the spinal cord: report of the two cases and review of the literature. Neurosurgery 1997; 41: 1410-1415. [ Links ]
12. BAYASIB Y, IWAYO M, HASEGAWA M. Malignan transformation of gangliocytoma/ganglioma into glioblastoma multiforme: a molecular genetic analysis, case report. J Neurosurg 2001; 95: 138-142. [ Links ]
13. BOEGLER O, SAWAYA R, RIVERA A, PELLOSKI C, SULMAN E. Prognostic and predictive markers in glioma and other neuroepithelial tumors. Curr Probl Cancer 2008; 32: 97-123. [ Links ]
14. PIETSCH T, TAYLOR M, RUTKA J. Molecular pathogenesis of childhood brain tumors. Journal of Neuro Oncology 2004; 70: 203-215. [ Links ]
15. GELABERT M, SERAMITO R, BANDIN J. Tumores talámicos bilaterales. Presentación de tres casos y revisión de la bibliografía. Rev Neurol 2007; 45: 599-603. [ Links ]
