










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Neurológica Colombiana
Print version ISSN 0120-8748
Acta Neurol Colomb. vol.27 no.3 Bogotá Sept./Nov. 2011
Utilidad de anticuerpos antiacuaporinas 4 en el diagnóstico de neuromielitis óptica
Usefulness of anti-aquaporine-4 antibodies in the diagnosis of neuromyelitis optica
Viviana Alexandra Martínez Villota. Residente Segundo Año Neurologia Clinica Universidad Nacional De Colombia. Medica Cirujana. Universidad Nacional De Colombia
Correo electrónico: halloviviana@yahoo.es
Recibido: 13/05/11. Revisado: 13/05/11. Aceptado: 18/06/11.
RESUMEN
La neuromielítis óptica o Enfermedad de Devic es un síndrome desmielinizante severo que a pesar de su baja incidencia, presenta un reto diagnóstico y terapéutico, principalmente en su diferenciación con esclerosis múltiple. Con el descubrimiento de la asociación de los anticuerpos contra canales de acuaporina 4 en la fisiopatología de este síndrome se han ampliado las herramientas para establecer un diagnóstico con mayor certeza y definir las posibilidades de tratamiento. A continuación se presenta una actualización de la literatura respecto al papel de los anticuerpos antiacuaporinas 4 en el diagnóstico y tratamiento de la neuromielítis óptica.
PALABRAS CLAVES. Neuromielítis Óptica, Acuaporina 4, Enfermedades Desmielinizantes, Neuritis Óptica, Mielitis Trasversa (DeCS).
SUMMARY
Neuromyelitis optica or Devic syndrome is a severe demyelinating syndrome. This entity although of low prevalence represents a diagnostic and therapeutic challenge, especially at the moment of differentiation with multiple sclerosis. With the discovery of aquaporin-4 specific serum autoantibody, in the pathophysiology of this syndrome, we have expanded the tools to make a diagnosis with greater certainty and identify options for treatment. Here is an update of the literature regarding its role in the diagnosis and treatment of neuromyelitis optica.
KEY WORDS. Neuromyelitis Optica, Aquaporin-4, Aquaporin-4 Specific Serum Autoantibody, Demyelinating Diseases, Optic Neuritis, Transverse Mielitis (MeSH).
INTRODUCCIÓN
La neuromielítis óptica (NMO), es un síndrome desmielinizante mediado por procesos autoinmunes, que inicialmente fue considerada parte del espectro de la esclerosis múltiple, sin embargo a partir de 1990 con las investigaciones sobre su fisiopatología, la asociación con los anticuerpos contra canales de acuaporina 4 y la relación entre varias enfermedades autoinmunes con este síndrome, se decidió posicio-narla como una patología diferente en el rango de los desórdenes autoinmunes del SNC.
A continuación se presenta una revisión de la literatura sobre la fisiopatología de la neuromielítis óptica asociada a anticuerpos antiacuaporinas 4, y la utilidad de éstos, en el diagnóstico y pronóstico de esta patología.
NEUROMIELÍTIS ÓPTICA
La neuromielítis óptica (NMO) fue descrita en 1870 por Sir Clifford Albutt y posteriormente en 1894 por Eugene Devic y Fernand Gault (1). Es un síndrome con múltiples etiologías y presentaciones clínicas, que constituyen el espectro diagnóstico de la neuromielitis óptica (Tabla 1). Afecta predominantemente el cordón espinal y el nervio óptico, simultáneamente o en recaídas, por lo cual su diagnóstico suele ser retador. Previamente se consideraba una forma de presentación clínica de Esclerosis Múltiple (EM), sin embargo el hallazgo de anticuerpos específicos antiacuaporinas y su asociación con otras patologías autoinmunes como Lupus Eritematoso Sistémico y Síndrome de Sjögren, la han ubicado en un rango diferente de los desordenes autoinmunes del sistema nervioso central, por lo tanto durante el presente texto, nos referiremos como neuromielítis óptica, a aquella asociada con anticuerpos antiacua-porina.

Epidemiología
El síndrome de NMO afecta principalmente a poblaciones con raza diferente a la blanca y con una menor contribución europea en su composición genética. Tiene una incidencia estimada menor a 1/100000 en países occidentales (1). En promedio inicia a finales de cuarta decada de la vida y la incidencia disminuye después de la quinta década. La mayoría de los pacientes sufren de un curso recurrente (80-90%), mientras que el curso monofásico (10-20%) o con progresión primaria o secundaria es raro. En la NMO monofásica los hombres y las muj eres se ven afectados por igual mientras que la forma recurrente es más frecuente en mujeres (80-90% con una razón 5-10:1). En 30% de los pacientes con curso monofásico y 23% del recurrente, se identificó una enfermedad viral previa. Como resultado de las lesiones acumuladas, la historia natural de la NMO se caracteriza por un deterioro progresivo de la función motora, sensorial, visual, intestinal y vesical (2).
FISIOPATOLOGÍA
La evidencia de mecanismos humorales en la patogénesis de NMO, llevó a la identificación de autoanticuerpos IgG específicos, ubicados en la barrera hematoencefálica (BHE), en la piamadre, el espacio de Virchow-Robin y microvasos en la sustancia blanca y gris del mesencéfalo, cerebelo y cordón espinal. Los anticuerpos se unen a un antígeno en la cara abluminal de los microvasos cerebrales, en un área representada por los procesos astrocíticos. Encontrar que los anticuerpos IgG-NMO se ubican en estas localizaciones, sugirió al canal de agua acuaporina 4 (AQP4) como el antígeno blanco en NMO (3). La AQP4 es un canal de agua impulsado por osmosis bidireccional que pertenece a la subfamilia de las acuaporinas de los mamíferos. En el SNC, la acuaporina 4 (AQP4) se expresa al pie de procesos astrocíticos alrededor de la membrana basal, en el nervio óptico, en una sub-población de células ependimarias en los núcleos del hipotálamo y en el órgano subfornical (4,5). En NMO el tercer bucle extracelular del AQP4 es considerado como el epítope importante para los anticuerpos contra AQP4. En los pies de los astrocitos el AQP4 se ubica junto al canal de potasio (Kir 4.1), que participa en la remoción de potasio extracelular. Esta localización sugiere que el flujo de agua y potasio está acoplados (6). También se asocia con el transportador de glutamato-1 (GLT-1), que impide la acumulación excesiva de glutamato extracelular (7). La localización estratégica de AQP4, junto con Kir 4.1 y GLT-1 ofrece varios posibles mecanismos por los cuales la pérdida de AQP4 puede provocar graves daños tanto en la mielina y los axones en las zonas vulnerables, así como edema reversible en otras regiones del cerebro, por ejemplo el hipotálamo y las estructuras periventriculares (8). La AQP4 perivascular permite un flujo de agua bi-direccional entre la sangre y el cerebro y se ha implicado en la patogénesis del edema cerebral (9). Se postula que una vez que los anticuerpos anti-AQP4 atraviesan la BHE, se unen a moléculas AQP4 y activan el complemento que moviliza neutrófilos y eosinófilos, para luego facilitar la destrucción del tejido. Los anticuerpos anti-AQP4 son subclase IgG1. La desmielinización es secundaria a la destrucción de los astrocitos, efecto que se supone fundamentalmente distinto del mecanismo desmielinizante primario ejecutado por la células T antígeno-específicas para proteína básica de mielina PBM y de anticuerpos anti mielina. Para inducir una recaída pueden requerirse otros factores que alteren la BHE.
Presentación clínica
Una de las principales preocupaciones es el diagnóstico diferencial de la NMO con esclerosis múltiple. Se ha descrito que en la neuritis óptica de la NMO, la pérdida visual suele ser más grave (10), es más probable una ocurrencia bilateral simultánea o secuencial en rápida sucesión (11) y en la oftalmoscopia se observa atrofia del disco más pronunciada que en la neuritis óptica por esclerosis múltiple. Otros hallazgos no difieren de la neuritis óptica por otras causas (12,13). En la NMO la ceguera se desarrolla en 60% de los pacientes con curso recurrente (media de seguimiento 16,9 años); y en el 22% de pacientes con curso monofásico (media de seguimiento 7,7 años) (7).
En NMO, el compromiso espinal se presenta como mielitis transversa completa con cuadripare-sia, un nivel sensorial casi simétrico y disfunción de esfínteres (2,14). Por el contrario, los síntomas en la esclerosis múltiple pueden ser más leves y asimétricos, una mielitis trasversa aguda parcial. El dolor radicular, los espasmos tónicos paroxísticos y el signo de L'hermitte se presentan en un tercio de los casos recurrentes de NMO, pero son raros en los pacientes con curso monofásico. Debido a la participación de centros de control medular de la respiración puede presentarse insuficiencia respiratoria que conduzca a la muerte (1). En la mielitis transversa de otras etiologías pueden presentarse alteraciones similares. En 15% de los pacientes con NMO, se presentan otras manifestaciones patológicas del sistema nervioso central; que incluyen encefalopatía, disfunción hipotalámica y deterioro cognitivo (15). También se han asociado endocrinopatías como amenorrea, galactorrea, diabetes insípida, hipotiroidismo e hiperfagia (16).
Diagnóstico
El diagnóstico es principalmente clínico, el apoyo de laboratorio clínico y neuroimágenes permiten establecer el diagnóstico diferencial y establecer si se encuentran patologías autoinmunes asociadas.Actualmente se consideran los anticuerpos antia-cuaporinas como el factor inmunológico más importante en su fisiopatología. Los criterios diagnósticos se resumen en la tabla 2.

Análisis de líquido cefalorraquídeo (LCR)
En el LCR se detectan anormalidades en la mayoría de los pacientes principalmente durante o poco después de un ataque agudo. Se encuentra pleocitosis en 14-79% de los pacientes que puede incluir o estar dominada por neutrófilos, el conteo celular es mayor de 50 células / ul en 13-35% de los pacientes y en algunos casos hasta 1000 células / ul. El aumento de los niveles de proteínas se halla en 46-75% de los casos. Se encuentran bandas oligoclo-nales en 0 - 37% y su presencia puede ser transitoria en la NMO en contraste con la EM (17). El valor de la determinación de neurofilamentos de cadena pesada (HNF) y proteína ácida glial fibrilar (GFAP) aún no se ha establecido claramente.
Evaluación electrofisiológica
Existen pocos estudios que evalúan específicamente la función electrofisiológica en pacientes con NMO. Un estudio cubano encontró potenciales evocados somatosensoriales anormales en 86% (42/49 pacientes), visuales en 83% (44/53 pacientes) y acústicos de tallo cerebral (BAEP) en 37% (19/51) (18). Las anormalidades de BAEP fueron más frecuentes en la raza negra (78% vs. 29% P = 0.003). La conducción nerviosa periférica motora y sensorial fue normal en los nueve pacientes con esclerosis múltiple ópticoespinal de un estudio japonés (19).
Hallazgos radiológicos
En la neuritis óptica (NO), la resonancia magnética por secuencias con supresión de grasa, (STIR, Short-tau inversion recovery) muestran aumento de la intensidad de señal en imágenes T2 del nervio óptico, en 84% de NO aguda y 20% durante la remisión en las neuritis ópticas en general. Se observó realce con contraste con gadolinio en secuencias spin echo T1 en NO aguda en 94% (20,21). No hay estudios que evalúen las diferencias en la resonancia magnética para la presentación de neuritis óptica en la NMO y la EM.
En la NMO, las lesiones de la médula espinal tienden a extenderse sobre tres o más segmentos vertebrales (Figuras 1 y 2 ). Se puede encontrar una apariencia de normalidad o menos lesiones, muy temprano en las recaídas o en la etapa atrófica residual (22). Las lesiones se observan como hiperinten-sidades en T2 e hipointensidades en T1 que suelen ocupar la mayor parte del área de sección transversal del segmento afectado y se asocian con inflamación y captación de gadolinio (23).
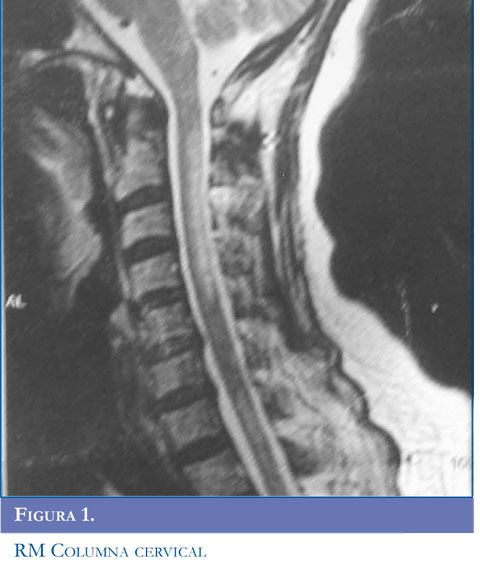
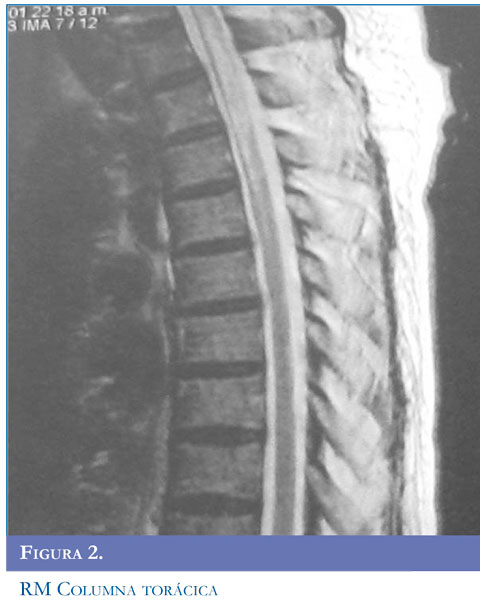
Inicialmente la RM del cerebro es normal, posteriormente pueden detectarse alteraciones hasta en 84,8% de los pacientes con NMO asociada a anticuerpos antiacuaporinas. La distribución de las lesiones cerebrales corresponde con áreas de alta expresión de AQP4 como las células ependimarias, el hipotálamo y tallo cerebral (24). La mayoría de las lesiones no son específicas puede ser asintomáticas y rara vez presentan lesiones que cumplan los criterios de Barkhof.
Anticuerpos antiacuaporinas
Los anticuerpos antiacuaporinas se diagnosticaron inicialmente mediante inmunofluorescencia indirecta (IFI), con una sensibilidad de 58 a 76% y especificidad del 85 - 99% para NMO. Se están estudiando otras técnicas incluidos los ensayos basados en: células (CBA), radioinmunoprecipitación, fluoroinmunoprecipitacion (FIPA) e inmunoabsor-ción ligado a enzimas (ELISA). Las sensibilidades y especificidades de los ensayos son diferentes y el patrón de oro está aún por dilucidar (25, 26).
Factores pronósticos
Los eventos índices son más severos en la forma monofásica de la NMO, que en la recurrente (11). Los eventos recurrentes causan deterioro clínico progresivo. Un mayor intervalo entre los dos primeros episodios clínicos, mayor edad de inicio, el sexo femenino y menor deterioro motor en el evento de mielitis centinela predicen un curso recurrente La historia de otras enfermedades autoinmunes, el aumento de la frecuencia de los ataques desmielini-zantes durante los 2 primeros años de la enfermedad y una mejor recuperación motora después del evento de mielitis índice, se asocian con un mayor riesgo de mortalidad en NMO recurrente. En un estudio basado en casos severos, 32% de pacientes con NMO recurrente fallecieron (media de seguimiento 60.2 meses) aunque no ocurrieron muertes en NMO monofásica. En otra cohorte se encontró 24 muertes entre 96 pacientes (25%), los predictores de mortalidad fueron: mayor frecuencia de ataques en el primer año de enfermedad, ceguera y alteración de esfínteres al inicio (27). En una cohorte de Brasil, con NMO recurrente, la mortalidad fue del 50% (28).
Valor de los anticuerpos antiacuaporinas 4 en la práctica clínica
Los anticuerpos antiAQP4 se han usado como predictores de recaída o de conversión a NMO después de un primer episodio de neuritis o mielitis. Se encontró que 62.5% de pacientes seropositivos con un episodio de mielitis trasversa extendida longitudinalmente, presentaron un segundo episodio dentro del siguiente año como mielitis o neuritis óptica, mientras que ninguno de los seronegativos recayó (29). Resultados iguales se observaron para neuritis óptica aislada recurrente con un mayor riesgo de conversión a NMO en el grupo con seropositividad (30, 35). Un estudio frances encontró que la presencia de IgG-NMO se asoció con recaídas más frecuentes, mayor ocurrencia de mielitis y mayor discapacidad relacionada con el ataque (31). La IgG-NMO se asocia con una mayor probabilidad de encontrar más de 3 lesiones periventriculares y localización en la profundidad de la sustancia blanca, y una lesión más extensa sobre la médula espinal durante la remisión. Los anticuerpos antiacuaporinas también se encontraron en trastornos del espectro de neuromie-lítis óptica: en mielitis trasversa longitudinalmente extendida LETM (50%) o neuritis óptica recurrente aislada RION (20%) (29, 35). Se ha encontrado que los niveles de anticuerpos séricos AQP4 se redujeron luego de tratamientos con rituximab, azatioprina, y ciclofosfamida (32), y metilprednisolona además se mantuvieron bajos durante el tiempo de la remisión.
Actualmente se recomienda que después de un primer episodio multifocal o monofocal, con seropo-sitividad para anticuerpos anti-AQP4, se debe iniciar un tratamiento inmunosupresor. Hasta un 30-40% de pacientes con un diagnóstico clínico bien establecido de NMO, persisten seronegativos para anti-AQP4, lo que se considera puede deberse a problemas de las técnicas o niveles de anticuerpos bajo el umbral de positividad de la prueba, además la inmunosupresión inducida por la terapia puede disminuir los niveles de anticuerpos y dar un resultado negativo en las pruebas. De otro lado, es probable que exista más de un antígeno blanco, un estudio identificó tres nuevos autoanticuerpos que están posiblemente implicados en la patogénesis de NMO (33). Además se debe considerar otros anticuerpos probablemente implicados, como en NMO asociadas a otras enfermedades autoinmunes como Lupus eritematoso sistémico o al síndrome de Sjógren. También la inmunidad celular puede tener importancia en algunos pacientes. Sin embargo, hasta ahora no hay evidencia de que los pacientes seronegativos deben tratarse de forma diferente a los seropositivos.
Tratamiento
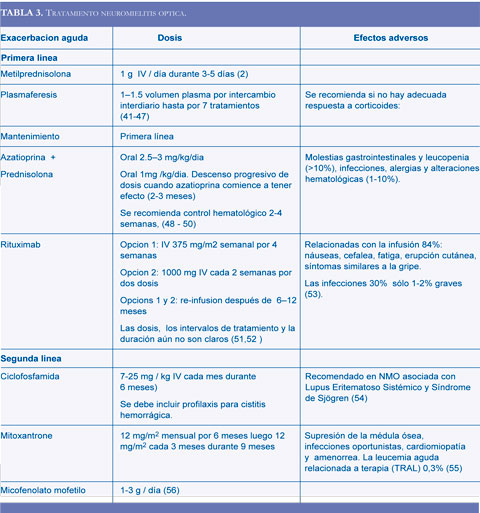
El tratamiento se divide en dos fases: aguda y mantenimiento. En la fase aguda se prefieren el uso de corticoides, aunque la plasmaféresis también ha dado buenos resultados. A largo plazo se recomienda azatioprina y corticoides, estudios iniciales con rituximab han mostrado buena efectividad, aún cuando las dosis y esquemas de tratamiento no se han establecido (Tabla 3).
CONCLUSIONES
En nuestro medio, donde los exámenes serológi-cos como la detección de anticuerpos anti-acuaporina 4 son difíciles de realizar por disponibilidad y costos, es importante elegir el paciente que se beneficiará de su realización. Un paciente con manifestaciones clínicas muy sugestiva como neuritis y mielitis cumple los criterios diagnósticos para NMO, los anticuerpos antiAQP4 pueden confirmar la patología para definir el inicio de tratamiento inmunosupresor a largo plazo, los potenciales efectos colaterales exigen estricto seguimiento.
Sin embargo, su mayor valor probablemente se encuentra en un paciente con cuadro clínicamente aislado de neuritis óptica o mielitis trasversa, dado que la alta especificidad de la prueba unida a un cuadro de alto riesgo puede confirmar la presencia de NMO, ya que estas presentaciones clínicas se aceptan actualmente como formas monofocales iniciales de NMO (34) y permiten por lo tanto, definir el inicio del tratamiento inmunosupresor en busca de disminuir las recurrencias y la discapacidad secundaria.
REFERENCIAS
1. SELLNER J, BOGGILD M, CLANET M ET AL. EFNS guidelines on diagnosis and management of neuromyelitis Optica. European Journal of Neurology 2010;17:1019-1032. [ Links ]
2. WINGERCHUK DM, HOGANCAMP WF, O_BRIEN PC, ET AL. The clinical course of neuromy-elitis optica (Devic_s syndrome). Neurology 1999;53:1107-1114. [ Links ]
3. LANA-PEIXOTO M. Devic's neuromyelitis optica. Arq Neuropsiquiatr 2008;66:120-138. [ Links ]
4. GRABER DJ, LEVY M, KERR D, ET AL. Neuromyelitis optica pathogenesis and aquaporin 4. J Neuroinflammation 2008;5:22-80. [ Links ]
5. TAIT MJ, SAADOUN S, BELL BA, ET AL. Water movements in the brain:role of aquaporins. Trends Neurosci 2008;31:37-43. [ Links ]
6. NAGGELHUS EA, HORIO Y, INANOBE A, ET AL. Immunogold evidence suggests that coupling of K+ siphoning and water transport in rat retinal Müller cells is mediated by a coenrichment of Kir4.1 and AQP4 in specific membrane domains. Glia 1999;26:47-54. [ Links ]
7. ZENG XN, SUN XL, GAO L, FAN Y, DING JH, HU G. Aquaporin-4 deficien-cy down-regulates glutamate uptake and GLT-1 expression in astrocytes. Mol Cell Neurosci 2007;34:34-39. [ Links ]
8. BENARROCH EE. Aquaporin-4, homeostasis, and neurologic disease. Neurology 2007;69:2266-2268. [ Links ]
9. GRIESDALE DE, HONEY CR. Aquaporins and brain edema. Surg Neurol 2004;61:34-39. [ Links ]
10. MERLE H, OLINDO S, BONNAN M, ET AL. Natural history of the visual impairment of relapsing neuromyelitis optica. Ophthalmology 2007;114:810-815. [ Links ]
11. WINGERCHUK DM, WEINSHENKER BG. Neuromyelitis optica:clinical predictors of a relapsing course and survival. Neurology 2003;60:848-853. [ Links ]
12. MANDLER RN, DAVIS LE, JEFFERY DR, ET AL. Devic_s neuromyelitis optica:a clinicopathologi-cal study of 8 patients. Ann Neurol 1993;34:162-168. [ Links ]
13. FARDET L, GENEREAU T, MIKAELOFF Y, ET AL. Devic_s neuromyelitis optica:study of nine cases. Acta NeurolScand 2003;108:193-200. [ Links ]
14. ORIORDAN JI, GALLAGHER HL, THOMPSON AJ, ET AL. Clinical, CSF, and MRI findings in Devic_s neuromielitis optica. J Neurol Neurosurg Psychiatry 1996;60:382-387. [ Links ]
15. POPPE AY, LAPIERRE Y, MELANCON D, ET AL. Neuromyelitis optica with hypothalamic involvement. Mult Sckr 2005;11:617-621. [ Links ]
16. VERNANT JC, CABRE P, SMADJA D, ET AL. Recurrent optic neuromyelitis with endocrinopathies:a new syndrome. Neurology 1997;48:58-64. [ Links ]
17. SEZE J, STOJKOVIC T, FERRIBY D, ET AL. Devic_s neuromyelitis optica:clinical, laboratory, MRI and outcome profile. J Neurol Sci 2002;197:57-61. [ Links ]
18. CABRERA-GOMEZ JA, KURTZKE JF, GON-ZALEZ-QUEVEDO A, ET AL. An epidemiological study of neuromyelitis optica in Cuba. J Neurol 2009;256:35-44. [ Links ]
19. KANZAKI M, MOCHIZUKI H, OGAWA G, ET AL. Clinical features of opticospinal multiple sclerosis with anti-aquaporin 4 antibody. Eur Neurol 2008;60:37-42. [ Links ]
20. JOHNSON G, MILLER DH, MACMANUS D, ET AL. STIR sequences in NMR imaging of the optic nerve. Neuroradiology 1987;29:238-245. [ Links ]
21. KUPERSMITH MJ, ALBAN T, ZEIFFER B, ET AL. Contrastenhanced MRI in acute optic neuritis:relationship to visual performance. Brain 2002;125:812-822. [ Links ]
22. WINGERCHUK DM, LENNON VA, PITTOCK SJ, ET AL. Revised diagnostic criteria for neuromy-elitis optica. Neurology 2006;66:1485-1489. [ Links ]
23. FILIPPI M, ROCCA MA, MOIOLA L, ET AL. MRI and magnetization transfer imaging changes in the brain and cervical cord of patients with Devic_s neuromyelitis optica. Neurology 1999;53:1705-1710. [ Links ]
24. PITTOCK SJ, LENNON VA, KRECKE K, ET AL. Brain abnormalities in neuromyelitis optica. Arch Neurol 2006;63:390-396. [ Links ]
25. FAZIO R, MALOSIO ML, LAMPASONA V, ET AL. Antiacquaporin 4 antibodies detection by different techniques in neuromyelitis optica patients. Mult Scler 2009;15:1153-1163. [ Links ]
26. WATERS P, VINCENT A. Detection of antiaquaporin-4 antibodies in neuromyelitis optica: current status of the assays. Int MS J 2008;15:99-105. [ Links ]
27. CABRE P, GONZALEZ-QUEVEDO A, BONNAN M, ET AL. Relapsing neuromyelitis optica:long term history and predictors of death. J Neurol Neurosurg Psychiatry 2009;80:1162-1164. [ Links ]
28. PAPAIS-ALVARENGA RM, MIRANDA-SANTOS CM, PUCCIONI-SOHLER M, ET AL. Optic neuromyelitis syndrome in Brazilian patients. J Neurol Neu-rosurg Psychiatry 2002;73:429-435. [ Links ]
29. WEINSHENKER BG, WINGERCHUK DM, VUKUSIC S, ET AL. Neuromyelitis optica IgG predicts relapse after longitudinally extensive transverse myelitis. Ann Neurol 2006;59:566-569. [ Links ]
30. DE SE-ZE J, ARNDT C, JEANJEAN L, ET AL. Relapsing inflammatory optic neuritis:is it neuromyelitis optica?. Neurology 2008;70:2075-2076. [ Links ]
31. MARIGNIER R, DE SEZE J, VUKUSIC S, ET AL. NMO-IgG and Devic_s neuromyelitis optica:a French experience. Mult Scler 2008;14:440-445. [ Links ]
32. JARIUS S, ABOUL-ENEIN F, WATERS P, ET AL. Antibody to aquaporin-4 in the long-term course of neuromyelitis optica. Brain 2008;131:3072-3080. [ Links ]
33. LALIVE PH, MENGE T, BARMAN I, CREE BA, GENAIN CP. Identification of new serum autoantibodies in neuromyelitis optica using protein microar-rays. Neurology 2006;67:176-177. [ Links ]
34. MARIGNIER R, GIRAUDON P, VUKUSIC S, CONFAVREUX C, HONNORAT J. Anti-aquapo-rin-4 antibodies in Devic's neuromyelitis optica:therapeutic implications. Ther Adv Neurol Disord 2010;3:311-321. [ Links ]
35. MATIELLO M, LENNON VA, JACOB A, ET AL. NMO-IgG predicts the outcome of recurrent optic neuritis. Neurology 2008;70:2197-2200. [ Links ]
36. WEINSHENKER BG, WINGERCHUK DM, VUKUSIC S, ET AL. Neuromyelitis optica IgG predicts relapse after longitudinally extensive transverse myelitis. Ann Neurol 2006;59:566-569. [ Links ]
37. PITTOCK SJ, LENNON VA, DE SEZE J, ET AL. Neuromyelitis optica and non organ-specific autoimmunity. Arch Neurol 2008;65:78-83. [ Links ]
38. MCKEON A, LENNON VA, JACOB A, ET AL. Coexistence of myasthenia gravis and serological markers of neurological autoimmunity in neuromyelitis optica. Muscle Nerve 2009;39: 87-90. [ Links ]
39. KIRA J. Neuromyelitis optica and asian phenotype of multiple sclerosis. Ann N Y Acad Sci 2008;1142:58-71. [ Links ]
40. MILLER DH, WEINSHENKER BG, FILIPPI M, ET AL. Differential diagnosis of suspected multiple sclerosis:a consensus approach. Mult Scler 2008;14:1157-1174. [ Links ]
41. LLUFRIU S, CASTILLO J, BLANCO Y, ET AL. Plasma exchange for acute attacks of CNS demy-elination:predictors of improvement at 6 months. Neurology 2009;73:949-953. [ Links ]
42. WEINSHENKER BG, OBRIEN PC, PETTER-SON TM, ET AL. A randomized trial of plasma exchange in acute central nervous system inflammatory demyelinating disease. Ann Neurol 1999; 46:878-886. [ Links ]
43. KEEGAN M, PINEDA AA, MCCLELLAND RL, ET AL. Plasma exchange for severe attacks of CNS demyelination:predictors of response. Neurology 2002;58:143- 146. [ Links ]
44. WATANABE S, NAKASHIMA I, MISU T, ET AL. Therapeutic efficacy of plasma exchange In NMO-IgG-positive patients with neuromyelitis optica. Mult Scler 2007;13:128-132. [ Links ]
45. RUPRECHT K, KLINKER E, DINTELMANN T, ET AL. Plasma exchange for severe optic neuritis:treatment of 10 patients. Neurology 2004;63:1081-1083. [ Links ]
46. BONNAN M, VALENTINO R, OLINDO S, ET AL. Plasma exchange in severe spinal attacks associated with neuromyelitis optica spectrum disorder. Mult Scler 2009;15:487-492. [ Links ]
47. MIYAMOTO K, KUSUNOKI S. Intermittent plasmapheresis prevents recurrence in neuromyelitis optica. Ther Apher Dial 2009; 13: 505-508. [ Links ]
48. MANDLER RN, AHMED W, DENCOFF JE. Devic_s neuromyelitis optica:a prospective study of seven patients treated with prednisone and azathio-prine. Neurology 1998;51:1219-1220. [ Links ]
49. BICHUETTI DB, LOBATO DE OLIVEIRA EM, OLIVEIRA DM, AMORIN DE SOUZA N, GABBAI AA. Neuromyelitis Optica Treatment:Analysis of 36 Patients. Arch Neurol 2010;67:1131-1136. [ Links ]
50. WINGERCHUK DM, WEINSHENKER BG. Neuromyelitis optica. Curr Treat Options Neurol 2005;7:173-182. [ Links ]
51. JACOB A, WEINSHENKER BG, VIOLICH I, ET AL. Treatment of neuromyelitis optica with rituximab:retrospective analysis of 25 patients. Arch Neurol 2008;65:1443-1448. [ Links ]
52. CREE BA, LAMB S, MORGAN K, ET AL. An open label study of the effects of rituximab in neuromyelitis optica. Neurology 2005;64:1270-1272. [ Links ]
53. KIMBY E. Tolerability and safety of rituximab (MabThera). Cancer Treat Rev 2005;31:456-473. [ Links ]
54. MOK CC, TO CH, MAK A, ET AL. Immunoablative cyclophosphamide for refractory lupus-related neuromyelitis optica. J Rheumatol 2008;35:172-174. [ Links ]
55. WEINSTOCK-GUTTMAN B, RAMANATHAN M, LINCOFF N, ET AL. Study of mitoxantrone for the treatment of recurrent neuromyelitis optica (Devic disease). Arch Neurol 2006;63:957-963. [ Links ]
56. JACOB A, MATIELLO M, WEINSHENKER BG, ET AL. Treatment of neuromyelitis optica with mycophenolate mofetil:retrospective analysis of 24 patients. Arch Neurol 2009;66:1128-1133. [ Links ]
