










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkActa Neurológica Colombiana
versão impressa ISSN 0120-8748
Acta Neurol Colomb. vol.28 no.1 Bogotá jan./mar. 2012
Gangliosidosis gml juvenil como causa de regresión en el neurodesarrollo: reporte de caso
Juvenile GM1 gangliosidosis as a cause of regression in neurodevelopment: a case report
Blair Ortiz, Servicio de Neurología Infantil, Universidad de Antioquia, Medellín. Claudia González, Eugenia Espinosa, Posgrado de Neurología Pediátrica, Hospital Militar Central, Universidad Militar Nueva Granada. Johana Guevara, Olga Yanet Echeverri, Luis. Barrera, Instituto de Errores Innatos del Metabolismo, Pontificia Universidad Javeriana, Bogotá..
Correo electrónico: blairortiz@hotmail.com
Recibido: 01/11/11. Revisado: 21/01/12. Aceptado: 13/02/12.
RESUMEN
La gangliosidosis GM1 es ocasionada por deficiencia en la actividad catalítica de la enzima lisosomal beta-galacto-sidasa, dando origen a la acumulación del esfingolípido conocido como gangliósido GM1. La enfermedad se manifiesta en forma generalizada, con trastornos neurológicos y visceromegalias. Reportamos un caso de presentación juvenil, masculino de 5 años con historia de pérdida de hitos del desarrollo motor, cognitivo y lenguaje en forma progresiva. Se diagnosticó gangliosidosis GM1 juvenil o tipo II luego del análisis de los estudios de laboratorio, neuroimágen y baja actividad enzimática de la beta-galactosidasa.
PALABRAS CLAVES. Gangliosidosis, GM1, Trastorno de Crecimiento, Regresión, Epilepsia, Leucoencefalopatías, Enfermedad por Depósito Lisosomal (DeCS).
SUMMARY
Gangliosidosis GM1 is due a deficiency of lysosomal acid beta-galactosidase which gives sphingolipids (GM1) accumulation. It has systemic compromise, mainly neurologic disease and organomegaly. Here, We report a 5-years old child with a juvenile presentation or type II, which is characterized by regression of neurodevelopment and progression to neurodegeneration. Based in his laboratory, neuroimaging and low enzymatic activity of beta-galactosidase a diagnosis of gangliosidoses GM1 was made.
KEY WORDS. Gangliosidosis, GM1, Growth Disorders, Epilepsy, Leukoencephalopathies, Lisosomal Storage Disease (MeSH).
INTRODUCCIÓN
La [3-galactosidasa es una enzima lisosomal involucrada en la degradación de glicoproteínas, glicosaminoglicanos como queratán sulfato y glicoes-fingolipidos, principalmente el gangliósido GM1; su déficit conlleva acumulación de estos compuestos en diversos tejidos. La deficiencia de esta enzima ocasiona dos entidades clínicas diferentes según el defecto genético presente, involucrando el sistema óseo o el sistema nervioso central (SNC). La primera entidad es el síndrome de Morquio B o mucopoli-sacaridosis IV B, en la cual se acumula el glicosami-noglicano queratán sulfato en el sistema óseo (1-3).
La gangliosidosis GM1 es una enfermedad de depósito lisosomal polisintomática, en ella destacan los trastornos neurológicos y aparición de viscero-megalias de grado variable, relacionado con el grado de actividad enzimática residual. Hay tres variantes clínicas: tipo I, infantil o de presentación neonatal: se manifiesta con trastorno de succión/deglución, hipoactividad, fascies toscas, visceromegalias y encefalopatía degenerativa severa. La tipo II o juvenil: tiene inicio después de los 6 meses de edad con retardo o involución del desarrollo psicomotor, trastorno de la marcha, pérdida del lenguaje, letargia, epilepsia y cuadriplejía espástica. La tipo III o del adulto: con inicio de los síntomas entre los 3 años y edad adulta, sin deterioro del neurodesarrollo, caracterizada por trastornos de la marcha, lenguaje y signos extrapiramidales de lenta progresión (1,3,4).
Presentación de caso. Escolar masculino de 5 años, hijo de padres no consanguíneos, llevado a la consulta de neurología pediátrica por presentar desde los 12 meses detención en el desarrollo psi-comotor, y 6 meses más tarde pérdida progresiva de los hitos motores y del lenguaje, desinterés por el medio y dificultad para la bipedestación. A los 4 años se evidenció pobre contacto con el medio e inició cuadro convulsivo con crisis focales secundariamente generalizadas (versión cefálica hacia la izquierda con postura tónica en extensión y movimientos clónicos de hemicuerpo izquierdo con generalización secundaria), se inició manejo con carbamazepina (CBZ), que fue remplazada posteriormente por levetirace-tam (LVT) por elevación de las transaminasas.
En el examen físico se encontró sin interacción con el medio, con perímetro cefálico en el percentil 15 (normal), con mirada fija con seguimiento visual ocasional, sin sostén cefálico, cuadriparesia espástica, reflejos músculo tendinosos exaltados en las 4 extremidades (3/4), Babinski bilateral, pie equino cavo bilateral, con sensibilidad táctil, dolorosa y profunda conservadas. Dependiente para todas las actividades de la vida diaria. Como antecedentes perinatales tuvo un embarazo y periodo perinatal sin complicaciones. Los antecedentes familiares no incluyeron enfermedades neuro-psiquiátricas ni genéticas.
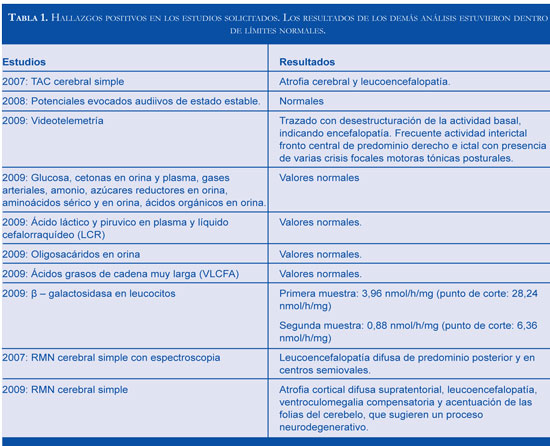
Durante los años 2007 y 2009 se realizaron análisis generales y especificos para trastornos meta-bólicos como: gases arteriales, pruebas de función hepática y renal, estudios metabólicos incluyendo determinación de actividad enzimática de arilsulfa-tasa A, hexosaminidasas, [3-galactosidasa y análisis de ácidos grasos de cadena muy larga en plasma. Además de otros estudios como electroencefalograma (EEG), electromiografía (EMG), tomografía axial computarizada (TAC) cerebral simple, resonancia magnética cerebral (RMC), videotelemetria EEG, potenciales evocados visuales y auditivos. La tabla 1 resume los hallazgos positivos.
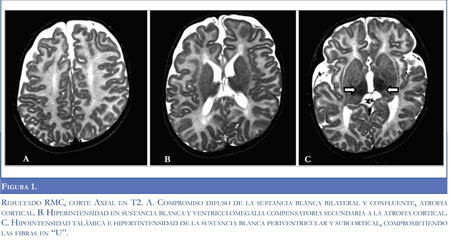
En la figura 1 se observan los resultados de la Resonancia Magnética Nuclear.
DISCUSION
El enfoque diagnóstico de las enfermedades neurodegenerativas de origen metabólico representa un reto para el personal medico debido a la alta heterogeneidad clínica observada, en especial en las formas de presentación tardía donde las manifestaciones neurológicas no son específicas.
La sospecha clínica de gangliosidosis GM1 está relacionada con la presencia de algunos características, que son más comunes en la forma infantil clásica tipo I que en la tipo II y III, entre los que se encuentran disostosis múltiple, facies toscas (nariz bulbosa, mejillas prominentes, labios gruesos y rosados), mancha rojo cereza en el fondo de ojo, hepatoesplenomegalia e historia de regresión en el desarrollo psicomotor (1,5). En ausencia de estos signos el diagnóstico se hace más difícil, como sucede en el caso presentado.
En el abordaje diagnóstico de un cuadro clínico de inicio en el lactante caracterizado por detención y posterior regresión en el neurodesarrollo, epilepsia, cuadriparesia espástica y leucoencefalopatía debe considerarse un amplio espectro de etiologías metabólicas, entre las que se encuentran las enfermedades de depósito lisosomal, peroxisomales y mitocondriales (5,6) (Figura 2). La confirmación de un caso como el presentado requirió de un detallado análisis de la evolución clínica del paciente y un amplio espectro de ayudas paraclínicas dentro de las que cobran especial importancia las imágenes cerebrales y baja actividad enzimática de la beta-galactosidasa (Tabla 1).
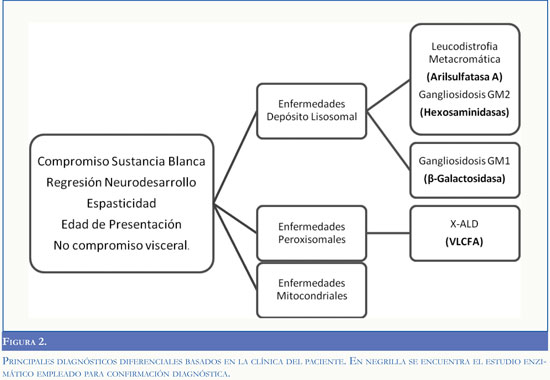
La leucodistrofia metacromática afecta clásicamente la sustancia blanca profunda sin lesionar las fibras subcorticales o fibras en "U"; mientras la adrenoleucodistrofia ligada al X (X-ALD) involucra la sustancia blanca profunda posterior. La determinación de la relación lactato/piruvato en plasma y LCR y de los niveles de VLCFA no mostraron alteración del metabolismo oxidativo mitocondrial ni peroxisomal, respectivamente.
La presencia de leucoencefalopatía difusa, con atrofia cortical y compromiso talámico observado en las imágenes de RMN es compatible con lo reportado en la literatura para gangliosidosis GM1 (7,8); estos hallazgos aunados a la historia clínica que sugerían una enfermedad neurodegenerativa y confirmación bioquímica de la deficiencia en la actividad de la beta-galactosidasa establecen el diagnóstico definitivo del paciente. Teniendo en cuenta el cuadro clínico y edad de presentación, se considera una variante juvenil o tipo II de gangliosidosis GM1. Los hallazgos de esta forma de presentación clínica son variables; los más frecuentemente reportados son retardo en el neurodesarrollo (96%), anormalidades esqueléticas (69%), facies toscas (66%), hipotonía (50%), epilepsia (18%) e hipertonía (4%) (1,6).
Actualmente no hay un tratamiento que remplace la enzima deficiente ni que supla por vía alterna la degradación del gangliósido GM1. El abordaje terapéutico consiste en el manejo de las complicaciones que presentan los pacientes, como el manejo de la espasticidad, epilepsia, postura, deambulación y los relacionados con la ingestión de líquidos y alimentos (1,3).
La gangliosidosis GM1 y los errores congénitos del metabolismo que ocasionan detención y regresión del neurodesarrollo son un reto para la comunidad científica porque requieren de un diagnóstico oportuno para informar y orientar adecuadamente a la familia.
REFERENCIAS
1. BRUNETTI-PIERRI N, SCAGLIA F. GM1 gangliosidosis: Review of clinical, molecular, and therapeutic aspects. Molecular Genetics and Metabolism 2008;94: 391-396. [ Links ]
2. MORITA M, SAITO S, IKEDA K, ET AL. Structural bases of GM1 gangliosidosis and Morquio B disease. J Hum Genet. 2009;54:510-515. [ Links ]
3. SUZUKI Y, NANBA E, MATSUDA J, HIGAKI K, OSHIMA A. p-Galactosidase Deficiency (P-Galactosidosis): GM1 Gangliosidosis and Morquio B Disease Part 16: LYSOSOMAL DISORDERS. Cap 151. En: Valle D, Beaudet A, Vogelstein B, KINZLER K, ANTONARAKIS S, BALLABIO A. The Online Metabolic and Molecular Bases of Inherited Disease. Mc Graw Hill. 2011. (http://www ommbid.com/). [ Links ]
4. MUTHANE U, CHICKABASAVIAH Y, KANESKI C, ET AL. Clinical features of adult GM1 gangliosi-dosis: report of three Indian patients and review of 40 cases. Mov Disord. 2004;19:1334-1341. [ Links ]
5. GASCON GG, OZAND PT, ERWIN RE. GM1 gangliosidosis type 2 in two siblings. J Child Neurol. 1992;7 Suppl:S41-50. [ Links ]
6. ROZE E, PASCHKE E, LOPEZ N, ET AL. Dystonia and parkinsonism in GM1 type 3 gangliosidosis. Mov Disord. 2005;20:1366-1369. [ Links ]
7. DE GRANDIS E, DI ROCCO M, PESSAGNO A, VENESELLI E, ROSSI A. MR imaging findings in 2 cases of late infantile GM1 gangliosidosis. AJNR Am J Neuroradiol. 2000;30:1325-1327. [ Links ]
8. EROL I, ALEHAN F, POURBAGHER MA, CANAN O, VEFA YILDIRIM S. Neuroimaging findings in infantile GM1 gangliosidosis. Eur J Paediatr Neurol. 2006;10:245-248. [ Links ]
