










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Neurológica Colombiana
Print version ISSN 0120-8748
Acta Neurol Colomb. vol.28 no.4 Bogotá Oct./Dec. 2012
Polineuropatía desmielinizante inflamatoria crónica. Aspectos clínicos y electrofisiológicos
CIDP: Clinical and electrophysiological aspects
Aymeé Hernández Hernández. Dra en Ciencias Médicas. Especialista de segundo grado en fisiología normal y patológica (Neurofisio-logía Clínica). Profesor Titular adjunto a la facultad de medicina Finlay-Albarrán de la Habana. Investigador Auxiliar. CCOI "Frank País". La Habana, Cuba. Correspondencia: aymeehh@infomed.sld.cu
Recibido: 05/07/12. Revisado: 18/07/12. Aceptado: 22/07/12.
RESUMEN
La polineuropatia desmielinizante inflamatoria crónica (CIDP), por sus siglas en inglés, es una polineurorradicu-lopatía predominantemente motora de etiología autoinmune. Su comienzo es insidioso y el curso crónico, afecta tanto a hombres como a mujeres con un ligero predominio en los primeros. Puede aparecer en cualquier etapa de la vida, es más frecuente a partir de la segunda década. Es necesario diferenciarla de otras enfermedades, ya que es potencialmente tratable, con buena respuesta a los inmunosupresores.
Su diagnóstico se sustenta en cuatro pilares: la clínica, los estudios neurofisiológicos, el estudio del líquido cefalorraquideo y los análisis anatomopatológicos. Desde su descripción inicial se han planteado numerosos criterios diagnósticos, aspecto polémico y controversial, ya que en la práctica clínica existen numerosos pacientes que no cumplen estrictamente con estos criterios pues presentan casos atípicos.
Por la dificultad en el diagnóstico de esta patología se realizó una revisión del tema, exponiendo los principales aspectos clínicos, electrofisiológicos, fisiopatológicos y anatomopatológicos, junto con una breve exposición de lo hallado en doce años de experiencia con esta patología en la Habana, Cuba.
PALABRAS CLAVES. PCID, Polineuropatía (DeCS).
SUMMARY
Chronic demyelinating inflammatory polineuropathy (CIDP) is a kind of peripheral neuropathy predominantly motor of autoimmune cause and chronic course. Its start insidiously, it can affect both sex, more frequent male. It can appear in any time of life more frequent in second decade. It is very important differentiate of other illness because it has good response to inmunosupresor agents.
Diagnosis is support ed in four aspects: clinical, electrodiagnostic studies, brain spinal fluid study and anatomo-pathologycal issues. Diagnosis is polemic and controversial, with different diagnosis criteria, because they not include atypical variants.
We have to review ed this topic, and show clinical, electrophysiological, fisiopathological and pathological aspects and also our experience in the study of CIDP patients throw twelve years in La Habana, Cuba.
KEY WORDS: CIDP, Polyneuropathies (MeSH).
INTRODUCCIÓN
La polineuropatía desmielinizante inflamatoria crónica (CIDP) al igual que el síndrome de Guillain-Barré (SGB) constituye una forma de afección del sistema nervioso periférico de causa autoinmune, con buena respuesta al tratamiento inmunomodu-lador e inmunosupresor. En su fisiopatología aún existen aspectos sin dilucidar (1,2).
A diferencia del síndrome de Guillain-Barré tiene un comienzo insidioso y un curso crónico, muchas veces no se diagnostica correctamente, y se confunde con otras afecciones del sistema nervioso periférico, de manera que empeora si el paciente no se trata adecuadamente.
Para su diagnóstico existen varios pilares: clínicos, electrofisiológicos y anatomo-patológicos. La clínica del paciente orienta, los estudios electrofisiológicos apoyan, y los anatomo-patológicos confirman el diagnóstico de esta afección.
Se han postulado, en varias ocasiones, criterios diagnósticos que tratan de facilitar el diagnóstico de la CIDP, sin embargo la mayoría de estos criterios dejan por fuera algunas formas poco comunes de presentación y se considera que en general son más útiles desde el punto de vista investigativo, no así en la práctica clínica ya que tienen baja sensibilidad (1-3).
Por todo lo anterior esta enfermedad resulta polémica y controversial, el hecho es que existe, afecta a un grupo considerable de pacientes, puede dejar secuelas invalidantes, si no se trata oportuna y adecuadamente e incluso puede ser expresión de alguna otra afección más compleja a la que en no pocas ocasiones se asocia.
Por ello se realizó una revisión sobre el tema y se muestra brevemente la experiencia del autor en doce años de atención a pacientes con la enfermedad.
La polineuropatía desmielinizante inflamatoria crónica (CIDP) es una polineurorradiculopatía mixta predominantemente motora, desmielinizante, inflamatoria, de comienzo insidioso, evolución crónica y etiología autoinmune (1-7).
También se ha denominado polineuropatía des-mielinizante inmune crónica, polirradiculoneropatía inflamatoria crónica, polineuropatía inflamatoria desmielinizante crónica, polineuropatía recurrente, polineuropatía dependiente de esteroides, neuritis hipertrófica (1- 3, 8, 9). Tiene una incidencia de 0.15 por 100 000 habitantes en el mundo con una prevalencia de 1 a 1.9 por 100 000. Su mortalidad es del 3% y genera secuelas en el 60% de los casos, el 20% son invalidantes (9).
Después de los reportes de Guillain y col., diferentes investigadores reportaron casos similares pero con un curso crónico y evolución recurrente. En 1958 Austin reportó 32 casos con una polineuropatía simétrica de curso fluctuante, estos casos mejoraron con la hormona ACTH.
Dyck y col. en 1968 estudiaron un número considerable de pacientes y demostraron que tenían disociación albúmino-citológica en el líquido cefalorraquídeo (LCR), que mejoraban con la terapia este-roidea, y propusieron pautas para el diagnóstico. En 1970 finalmente emergió la CIDP como una entidad distinta al SGB; con características clínicas, histopa-tológicas y electrodiagnósticas definidas. Numerosos investigadores han estudiado series de casos y postulando criterios diagnósticos (como Barohn y col en 1989), los cuales han evolucionado hasta los actuales postulados por el mismo Barohn en el año 2000, pasando por los de la Academia Americana de Neurología Ad hoc subcommittee (1, 3, 8, 10, 11).
Síntomas y signos
La CIDP puede aparecer a cualquier edad, aunque es más frecuente entre los 30 y los 50 años. Se han reportado casos con inicio en la infancia e incluso formas congénitas con remisión espontánea, aunque son poco frecuentes. También se han descrito casos frecuentes relacionados con el transplante de órganos. La razón hombre/mujer es: 2:1.
Tiene un comienzo insidioso con debilidad muscular simétrica, fundamentalmente proximal, calambres, parestesias, dolor en las extremidades con predominio de las inferiores por donde suelen comenzar los síntomas. Es infrecuente el compromiso de pares craneales, el séptimo par es el más afectado. Pueden existir atroia muscular, fasciculaciones, disminución o pérdida de los reflejos osteotendino-sos, engrasamiento de nervios y temblor intencional (1, 4-7, 12,13). Puede evolucionar de varias formas: progresiva crónica, estacionaria, con empeoramiento escalonado y con recaídas (1, 4-7, 12-14, 15).
Criterios diagnósticos
Para la CIDP se han postulado criterios diagnósticos que tienen utilidad con fines investigativos, a los mismos se le han realizado varias modificaciones a lo largo del tiempo, tratando de aumentar su sensibilidad (9,14,15).
Los primeros criterios fueron expuestos por Dyck y colaboradores en 1975, incluyendo la debilidad muscular simétrica, tanto proximal como distal de más de 6 meses de evolución, aumento de las proteínas en el LCR; así como estudio de conducción nerviosa (ECN) y biopsia de nervio sural que demuestren desmielinización (16).
En 1989 Barohn y colaboradores expusieron como criterios: la arreflexia o hiporreflexia marcada, debilidad muscular proximal y distal simétrica en extremidades superiores e inferiores, evolución de más de dos meses, aumento de las proteínas en el LCR, ECN y biopsia que demuestren desmielinización, y se aceptó la CIDP asociada a enfermedades concurrentes. Estos criterios usaron un enfoque minimalista del ECN con signos de desmielinización (sólo planteó la disminución de la velocidad del conducción motora (VCNM) menor del 70% en dos nervios) (17).
Posteriormente en 1991 Cornblath y el Ad Hoc subcomittee de la AAN propusieron unos criterios umversalmente aceptados:
Clínicos: polineuropatía simétrica que afecta fibras sensitivas y motoras, con predominio de estas últimas; debilidad muscular simétrica progresiva proximal y distal, de extremidades superiores e inferiores; hipo o arreflexia osteotendinosa en más de una extremidad y progresión del cuadro de más de 2 meses.
De laboratorio: aumento de las proteínas en el LCR mayor de 0.45mg/L con células menores de 10 /mm3. Estudio de conducción nerviosa con características de desmielinización: velocidad de conducción nerviosa disminuida en al menos 2 nervios motores menor o igual al 70-80% del límite inferior de normalidad, latencia distal prolongada en 2 o más nervios mayor o igual al 125-150% del límite superior de normalidad. Bloqueo parcial de la conducción motora: aumento de la caída de amplitud pico-pico mayor o igual al 20% con dispersión mayor o igual al 15% o dispersión temporal anormal en uno o más nervios (aumento de la caída de amplitud pico-pico mayor o igual al 20% con dispersión mayor o igual al 15%). Alteración de la onda F: ausencia o prolongación en 2 o más nervios mayor o igual al 120-150% del límite superior de normalidad. Enlentecimiento de la velocidad de conducción nerviosa sensitiva menor o igual al 80% del límite inferior de normalidad y ausencia de reflejo H. Biopsia de nervio característica con desmielinización: evidencias inequívocas de desmielinización y remielinización, desmielinización segmentaria, formación de bulbos de cebolla, variación del grado de desmielinización entre los fascículos, pérdida de fibras nerviosas, inflamación perivascular, signos de remielinización, edema endoneural o subperineural, infiltrado de células mononucleares.
Criterios de exclusión. En la clínica neuropatía sensorial pura, ictiosis, retinitis pigmentaria, exposición a drogas, tóxicos o toxinas, neuropatías congénitas, alteraciones esfinterianas, nivel sensitivo. En el laboratorio: niveles bajos de colesterol, metabolitos porfirínicos, disminución de vitamina B12, intoxicación por metales pesados, aumento de la glucosa, aumento de la celularidad del LCR. En la biopsia: elementos que indiquen vasculitis, vesículas neurofilamentosas en los axones, depósitos amiloides o de enfermedades típicas como la de Fabry, leuco-distrofia metacromática, enfermedad de Refsum, etc. En el electrodiagnóstico: características que apunten a otras enfermedades como: defecto en la transmisión neuromuscular, miopatía o enfermedad de motoneurona.
Se dice que la CIDP es definida: cuando hay criterios clínicos, electrodiagnósticos y estudio del LCR positivos, que es probable: con criterios clínicos, electrodiagnósticos y biopsia positivos o criterios clínicos, LCR y biopsia positivos. Es posible: con criterios clínicos positivos y uno de de los tres criterios de laboratorio positivos (15).
Estos criterios dieron énfasis a los elementos electrodiagnósticos, detallaron los parámetros elec-trofisiológicos para la desmielinización (incluyeron la onda F y la latencia distal), desestimaron el requisito de aumento de las proteínas en el LCR así como la presencia de debilidad muscular simétrica y plantearon que los criterios solo deben emplearse como una herramienta de investigación (15).
En 2000 Barohn dio a conocer otros criterios: debilidad muscular simétrica, tanto próximal como distal con más de 2 meses de evolución, en extremidades superiores e inferiores, aumento de las proteínas en el LCR, electrofisiología que evidencia desmielinización; que desenfatizan la necesidad de la biopsia de nervio, evidencia de desmielinización sólo en la mitad de los pacientes, inflamación en el 10%, el nervio parece normal en el 20%, así como proteínas en el LCR (18).
Nuevamente, en 2001 Saperstein expuso nuevos criterios para el diagnóstico de la CIDP: con simetría y enfasis en la afectación próximal, los criterios elec-trofisiológicos son similares a los de la AAN, excepto que la definición del bloqueo fue más restringida y sólo se necesitan 2 criterios, la biopsia no se necesita para la definición, aunque es importante para la categoría de probable y sí el estudio del LCR (15).
En 2001 el Hughes and Inflammatory Neuropathy Cause and Treatment Group (INCAT) expuso sus criterios. En lo clínico: alteración motora y sensitiva de más de una extremidad mayor de 2 meses y arreflexia o hiporreflexia marcada. En el electrodiagnóstico: bloqueo parcial de la conducción o dispersión temporal anormal en al menos 2 nervios (caída de la amplitud mayor al 20% con cambios en la duración). Disminución de la VCN (menor del 70-80% de lo normal), prolongación de la latencia distal (superior al 125%) y ausencia de onad F o una latencia superior al 120%. En presencia de anomalías neurofisiológicas significativas en solo 2 nervios deben existir evidencias histológicas inequívocas de desmielinización. Apoyan el diagnóstico con aumento de las proteínas y células normales el LCR (19).
Estos criterios son menos restrictivos que los de la AAN y los de Saperstein ya que solo necesitan 3 anormalidades para evidenciar desmielinización y ellas incluyen: bloqueo parcial o dispersión temporal anormal, latencia de onda F, latencia distal o VCN, y requieren la evaluación de segmentos proximales. Su desventaja es que considera para el diagnóstico de CIDP que en el nervio ocurre la combinación de pocas anormalidades específicas (19).
En 2002 Nicolas y colaboradores expusieron unos criterios puramente electrodiagnósticos: bloqueo parcial de la conducción (caída de la amplitud en más del 30%) o dispersión temporal anormal (cambios en la duración superior al 15%) en al menos 3 nervios con conducción anormal en 1 nervio. Bloqueo parcial de la conducción o dispersión temporal anormal en al menos 2 nervios con conducción anormal en 1 nervio o bloqueo parcial de la conducción o dispersión temporal anormal en al menos 1 nervio con conducción anormal en 2 nervios. Sin bloqueo parcial, pero con conducción anormal en 3 nervios.
Estos criterios incluyen los sitios más proximales de estimulación, tales como el punto de Erb, y son más restrictivos en cuanto a la deinición del bloqueo parcial de la conducción (20).
También en 2002 Thaisetthawatkul y colaboradores propusieron otros criterios, agregando la prolongación de la duración distal motora igual o mayor a 9 ms, lo cual mejora la sensibilidad diagnóstica al 78% (21).
Por último en 2005 el Joint Task force EFNS and PNS expuso sus criterios para el diagnóstico de CIDP como típica sí existía: debilidad muscular proximal y distal, o atípica: con DADS, pura motora/sensitiva, síndrome de Lewis-Sumner, focal (plexopatía), participación del sistema nervioso central. La presencia de elevación de las proteínas en el LCR con menos de 10 células, RMN con hallazgos de adelgazamiento de raíces nerviosas, biopsia de nervio con evidencia de desmielinización o mejoría clínica con terapia inmunomoduladora apoyan el diagnóstico. Criterios de exclusión: neuropatías hereditarias, antecedentes de difteria o exposición a otras toxinas, alteraciones esinterianas, neuropatía motora multifocal (NMM) o anti-MAG (22).
Éstos criterios requieren la presencia de des-mielinización en dos nervios para la deinición, son más flexibles al cambiar la definición de probable, en cuanto a la caída de amplitud de 50 a 30%, la categoría de posible requiere solo de la afectación de un nervio (no menciona sobre los sitios de atrapamiento) (22).
Como se habrá apreciado los criterios para el diagnóstico de la CIDP resultan polémicos y con-troversiales. Los criterios del subcomité ad hoc y de la AAN son útiles para diagnosticar solamente el 60% de los pacientes con CIDP. El diagnóstico correcto puede hacerse si hay evidencia clara de desmielinización primaria por los resultados de los estudios electrofisiológicos o anatomopa-tológicos. Los criterios diagnósticos tienen una sensibilidad de 56-70% y una especificidad de 85-98% (23, 24).
Se plantea que los criterios de AAN tienen una sensibilidad del 40%, los de Saperstein del 47%, los de Nicolas del 53%, los de INCAT 60% y los de Thaisetthauakul del 70% (24).
Las formas clínicas aparecen en la tabla 1. Forma sensitiva pura. Es un término controversial, muchos autores plantean que es un estado clínico inicial, que precede a la debilidad en el 70% de los casos. En ella se produce alteración sensitiva con función motora normal o con mínima debilidad distal. Hay poca respuesta a los esteroides y se ha observado mejoría del cuadro en respuesta al tratamiento con inmunoglobulina humana endovenosa (IgIV). En otros casos hay afección de fibras mielinizadas gruesas de las raíces dorsales, produciéndose ataxia; afección de raíces sensitivas, con aumento de las proteínas en el LCR, la respuesta sensitiva es normal en el estudio de conducción. En la biopsia de raíces sensitivas se evidencia adelgazamiento de las mismas, disminución de la densidad de ibras mielinizadas gruesas, desmieli-nización segmental, formaciones de bulbos de cebolla, inflamación endoneural (9, 25, 26).

Forma infantil La CIDP es rara en los niños, en el 54% de los casos se asocia a infección previa o vacunas; el cuadro clínico es similar al de los adultos, al igual que las alteraciones del LCR; el estudio electroisiológico no cumple los criterios en todos los casos; el inicio es más abrupto que en los adultos y responden bien al tratamiento inmunomodulador (27, 28).
Formas axonales. En estos casos los estudios electroisiológicos y la histopatología sugiere una axonopatía primaria, el estudio del LCR tiene las características de la CIDP, responde bien a la terapia inmunosupresora (29-31).
Forma asociada a otras entidades. Se plantea que en el 12% de los casos la CIDP se asocia a otra enfermedad (estas fueron enumeradas en la clasiicación de las variantes). Responden menos al tratamiento habitual para la CIDP (29-31).
Eventos que pueden anteceder a la CIDP. A diferencia del SGB, en la CIDP por lo general no se asocian antecedentes importantes relacionados con el inicio del cuadro neurológico, aunque se han reportado casos de CIDP relacionados con infección por virus de la hepatitis C. En la forma infantil se plantea que existe asociación con infecciones precedentes y vacunas (27, 28).
Fisiopatología de la CIDP. La fisiopatología exacta no está mejor establecida que en otras neuropatías autoinmunes, ya que los factores que desencadenan la CIDP no se han podido establecer con exactitud. Se sabe que ocurre una respuesta inmune con producción de anticuerpos del tipo IgG e IgM que actúan contra los gangliósidos GM1, GD1a, GD1b, GT1b, LM1, sulfátidos y tubulinas del citoesqueleto. Existen productos de activación del complemento, participación de linfocitos T CD4 y CD8, con acción de linfoquinas pro inflamatorias: citoquinas, interleukinas 1 y 6, interferón y macro-fagos, todo lo cual produce lisis focal de la mielina (9, 32-39).
Las alteraciones histopatológicas pueden conir-marse en la biopsia de nervio sural (38-40). Diagnóstico diferencial. El diagnóstico diferencial de la CIDP debe establecerse con entidades tales como: Síndrome de Guillain Barré, neuropatías hereditarias, neuropatías tóxicas, enfermedad de la motoneurona, neuropatías que acompañan a procesos malignos como las neoplasias y las ganmapatías monoclonales, neuropatía motora multifocal (1, 2, 21, 41, 42).
TRATAMIENTO
Al igual que en el SGB no se conoce forma de evitar la afección. En el tratamiento de la entidad se postulan entre otros: la plasmaféresis, la inmunoglo-bulina, los esteroides y los citostáticos (43-51). En relación con la plasmaféresis la cronicidad del cuadro clínico, hace que se prolongue el tratamiento y limita su uso (43, 45-47, 51). La inmunoglobulina humana endovenosa (IgIV) se usa en ciclos mensuales o trimestrales en IgIV dosis de 0.4 gramos por kg/día durante 5 días (43-47, 50, 51). Produce inactivación del sistema de complemento, acción sobre los linfocitos T CD4 y CD8, compitiendo con el reconocimiento de los antí-genos, posee acción sobre los linfocitos B, disminuyen la producción de anticuerpos, bloquea al receptor Fc, produciendo interferencia en la fagocitosis mediada por receptor, impide la acción de los autoanticuerpos, inactiva los autoanticuerpos circulantes, disminuye la producción de citocinas y las inactiva, además estimula la remielinización (44, 50,51).
Los esteroides se utilizan con dosis de hasta 120 mg diarios por vía oral. Suprimen la respuesta inflamatoria, disminuyen la acción de los autoanti-cuerpos y las células T, (43, 45-48). Los citostáticos se reservan para los pacientes que no han tenido una respuesta satisfactoria a inmunoglobulina humana endovenosa, a la plasmaféresis o al tratamiento este-roideo. Entre ellos los más usados son: la azatioprina 2-3 mg kg/día, la ciclofosfamida 1.5-2 mg kg/día y la ciclosporina 3-6 mg kg/día (48, 49).
Se ha reportado el uso de interferón aunque los resultados no superan el del uso de la IgIV y los esteroides (45-51).
RESUMEN DE EXPERIENCIA DURANTE 12 AÑOS
Aspectos clínicos. Entre enero de 1999 y mayo de 2012 se evaluaron 34 pacientes con diagnóstico clínico y anatomopatológico de CIDP. Los que procedían de la consulta y sala de hospitalización de neurología del hospital "Carlos J. Finlay", en la Habana, Cuba.
De ellos 28 fueron hombres y 8 mujeres (3,5:1), con edades comprendidas entre 20 y 80 años, con una media de 52.3 años. El tiempo de evolución de la enfermedad osciló entre 21 días y 7 años, con una media de un año y medio.
De los pacientes evaluados 24 presentaron CIDP primaria o idiopática y 10 presentaron CIDP asociada a otras enfermedades; de este segundo grupo: siete presentaron CIDP asociada a diabetes mellitus, un paciente presentó CIDP asociada a hipertiroidismo, un paciente presentó CIDP asociada a infección por VIH, un paciente presentó CIDP asociada a esclerosis múltiple y un paciente presentó CIDP asociada a adenocarcinoma de vejiga.
El diagnóstico inicial estuvo errado en doce de estos pacientes: en un paciente se pensó en una neuropatía motora multifocal por presentar inicial-mente un cuadro asimétrico y toma del cuarto par craneal, en otro paciente se pensó en una plexopatía braquial izquierda por presentar debilidad progre siva del miembro superior izquierdo con plejía, en tres pacientes se pensó inicialmente en una radiculopatía S1 por presentar pérdida de la fuerza muscular y trastornos parestésicos en dicho territorio, sin dolor. En los siete pacientes con diabetes se pensó en una polineuropatía diabética.
El 100% de los pacientes presentó síntomas y signos motores y sensitivos, predominando los primeros. El 59.2% de los pacientes presentó atrofias e hipotrofias musculares, coincidiendo con mayor tiempo de evolución de la enfermedad. La afección de pares craneales solo se observó en dos casos (7.4%).
A este grupo de pacientes se le realizó evaluación neurofisiológica que incluyó estudio de conducción nerviosa periférica motora por tramos y sensitiva de los principales nervios de las cuatro extremidades, así como onda F de los nervios mediano y tibial posterior bilateralemente. Los resultados de estos estudios apoyaron el diagnóstico de CIDP, observando alteraciones incluso en los miembros aparentemente sanos; dicho diagnóstico fue confirmado a través del estudio anatomo-patológico, en biopsia de nervio sural.
Evaluación neurofisiológica
El estudio sensitivo mostró prolongación de la latencia, de la duración y VCN, así como disminución de la amplitud de las respuestas sobre todo en el estudio del nervio sural, lo que implica daño axonomielínico más intenso del nervio; no cumpliéndose en la mayoría de los casos (81.48% de los nervios evaluados) el patrón de anormalidad de la amplitud del estudio sensitivo del nervio mediano/ normalidad en el sural planteado por otros autores (52-58). Este patrón sí se cumplió para la mayoría de pacientes con SGB (60% de los nervios), evaluados en otro estudio realizado por Hernández en 2008 (59). Este es un punto de diferencia importante entre ambas entidades.
En el estudio motor las respuestas presentaron latencias y duraciones prolongadas, amplitud y área disminuidas y VCN enlentecida, así como mayor porcentaje de caída de amplitud, de área y dispersión (estas tres últimas variables definen el bloqueo parcial de la conducción, típico de esta enfermedad). La onda F presentó prolongación de la latencia media. Además gran parte de las respuestas fueron poco replicables y con morfología anormal.
Además se realizaron potenciales evocados somatosensoriales a la mitad de los casos evaluados observando positividad de los mismos en el 76% de los pacientes evaluados, con prolongación de la latencia de la onda P40 de forma bilateral en todos los casos, lo que sugiere la ocurrencia de desmieli-nización también en estructuras centrales, planteada por otros autores.
Se realizó la comparación de las medias de las variables neurofisiológicas analizadas correspondiente a los pacientes con CIDP primaria y secundaria, a través de una t-student para muestras independientes, se demostró la existencia de diferencias estadísticamente significativas entre las variables electrofisiológicas de los pacientes con CIDP primaria y secundaria.
En el estudio sensitivo los pacientes con CIDP primaria mostraron latencias y duraciones más prolongadas y VCN más lenta (Figura 1). En el estudio motor los pacientes con CIDP secundaria mostraron respuestas con mayor disminución de la amplitud y del área, VCN más lenta y mayor porciento de caída de amplitud y de área en comparación con los que presentaron CIDP primaria, predominando la afectación motora (Figura 2). La onda F de pacientes con CIDP secundaria presentó mayor prolongación de la latencia que los pacientes con CIDP primaria.
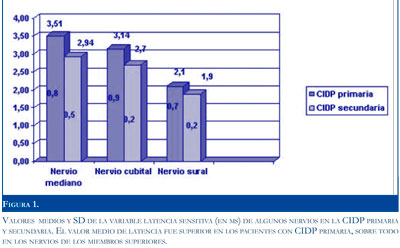
Las alteraciones de los estudios electrofisiológicos fueron más intensas en los pacientes con CIDP secundaria, traduciendo mayor daño axono-mielínico del nervio en este último grupo de pacientes.
CONCLUSIONES
La CIDP es una polineurorradiculopatía predominantemente motora de etiología autoinmune con aspectos polémicos y controversiales en su diagnóstico.
Los estudios neurofisiológicos constituyen un pilar importante para su diagnóstico. Existe diferente comportamiento electrofisioló-gico entre las variantes de la CIDP. La intensidad de alteración en los estudios es mayor en pacientes con CIDP secundaria.
REFERENCIAS
1. GUTIÉRREZ RIVAS E, JIMÉNEZ MD, PARDO J, ROMER J, EDS. Manual de Electromiografía Clínica. Barcelona España: Prus Science. 2000. [ Links ]
2. DYCK PJ, THOMAS PK. Peripheral Neuropathy 3 Ed. New York. WB Saunders. 1993;2:1298-1524. [ Links ]
3. BUSBY M, DONAGHY M. Chronic dysimmune neuropathy. A subclassification based upon the clinical features of 102 patients. J. Neurology. 2003;250:714-24. [ Links ]
4. SAPERSTEIN DS, KATZ JS, AMATO AA, BAROHN RJ. Clinical spectrum of chronic inflammatory demyelinating polineuropathy. Muscle Nerve. 2001;24:311-24. [ Links ]
5. KATZ JS, SAPERSTEIN DS, AMATO AA, BAROHN RJ. The clinical spectrum of chronic inflammatory demyelinating polineuropathy. 1KP. 002.AAN. Syllaby. 2000. [ Links ]
6. AMATO AA. Chronic inflammatory demyelinating polineuropathy. 7FC.002. AAN Syllaby 2000. [ Links ]
7. PASCUAL PASCUAL SL. Aspectos actuales de las neuropatías inflamatorias agudas y crónicas. Síndrome de Guillain-Barré y polineuritis crónica inflamatoria desmielinizante. Rev Neurol. 2002;35:269-76. [ Links ]
8. ASBURY A. Approach to the patient with peripheral neuropathy in Harrisons Principles of Internal Medicine. 15 Ed. New York. Mc Graw-Hill, 2001. [ Links ]
9. SERRADEL AP. Neuropatías disinmunes adquiridas. Sintomatología clínica y clasificación. Rev Neurol. 2000;30:501-14. [ Links ]
10. GRIFFIN JW. The Guillain Barre syndrome and chronic demyelinating inflammatory polyneuropathy 8BS002; AAN Syllaby. 1998. [ Links ]
11. OH SJ, KUROKAWA K, ALMEIDA DF, RYAN E, CLAUSSEN C. Subacute inflammatory demyelinating polineuropathy. Neurology. 2003;61:1507-12. [ Links ]
12. VALLAT JM, TABARAUD F, MAGY L, MACIAN F. Chronic polyradiculoneuritis and its frontiers. Rev Neurol (Paris). 2002;158: 27-31. [ Links ]
13. BURNS TM. Chronic inflammatory demyelinating polyradiculoneuropathy. Arch Neurol. 2004;61:973-5. [ Links ]
14. ADAMS RD, VICTOR M, ROPPER AH. Principles of Neurology. 6 Ed. México. Mc Graw-Hill Interamericana. 1999;1530-45. [ Links ]
15. CORNBLATH DR, ASBURY AK, ALBERS JW. Ad hoc subcommittee of AAN AIDS task force: Research criteria for diagnosis of chronic inflammatory demyelinating polineuropathy. Neurology. 1991;41:617-8. [ Links ]
16. DYCK PJ, LAIS AC, OHTA M. Chronic inflammatory polyradiculoneuropathy. Mayo Clin Proc. 1975;50:621-37. [ Links ]
17. BAROHN RJ. Clinical approach to peripheral neuropathy. 2FC.002; AAN Syllaby. 1998 [ Links ]
18. CORNBLATH DR, ASBURY AK, ALBERS JW. Ad hoc subcommittee of AAN AIDS task force: Research criteria for diagnosis of chronic inflammatory demyelinating polineuropathy. Neurology. 1991;41:617-18. [ Links ]
19. BAROHN RJ, LOIS CA, SMITH DE. Criterios diagnósticos del año 2000 para la polineuropatía des-mielinizante inflamatoria crónica. Boletín de noticias del síndrome de Guillain-Barré, Verano. 2000:1-3. [ Links ]
20. HUGHES R, BENSA S, WILLISON H. ET-AL. Ramdomized controled trial of intravenous immunoglobulin versus oral prednisolone in chronic inflammatory demyelinating polyradiculoneuropathy. Ann Neurol. 2001;50:195-201. [ Links ]
21. NICOLAS G, MAISONOBE T, LE FORESTIER N, LÉGER JM, BOUCHE P. Proposed revised electrophysiological criteria for chronic inflammatory demyelinating polyradiculoneuropathy. Muscle Nerve. 2002; 25:26-30. [ Links ]
22. THAISETTAHWATKUL P, LOGIGIAN E, HERRMANN DN. Dispersión of the distal compound muscle action potential as a diagnostic criterion for chronic inflammatory demyelinating polyneuropathy. Neurology. 2002;59:1526-32. [ Links ]
23. LEWIS RA. Therapy of GBS and CIDP. Conferencia en el 3er Encuentro de enfermedades neuro-musculares y visuales en la Habana, Febrero de 2006. [ Links ]
24. MAGDA P, LATOV N, BRANNAGAN TH, WEIMER LH, CHIN RL, SANDER HW. Comparison of electrodiagnostic abnormalities and criteria in a cohort of patients with chronic inflammatory demyelinating polirradiculoneuropathy. Arch Neurol. 2003;60:1755-9. [ Links ]
25. MOLENAAR DSM, VERMEALEN M, DE HAAN RJ. Comparison of electrodiagnostic criteria for demyelination in patients with chronic inflammatory demyelinating polyneuropathy (CIDP). J. Neurology. 2002;249:400-3. [ Links ]
26. VAN DIJK GW, NOTERMANS NC, FRANS-SEN H, WOKKIE JHJ. Development of weakness in patients with Chronic inflammatory demyelinating polyneuropathy and only sensory symptoms at presentation: A long-term follow-up study. J. Neurology. 1999;246:1134-9. [ Links ]
27. SABATELLI M, MADIA F, MIGNONA T, LIPPI G, QUARANTA L. Pure motor chronic inflammatory demyelinating polyneuropathy. J. Neurology. 2001;248:772-7. [ Links ]
28. SIMMOUNS Z, WALD JJ, ALBERS JW. CIDP in children: Presentation, electrodiagnostic studies and initial clinical course with comparison to adults. Muscle Nerve. 1997;20:1008-15. [ Links ]
29. MAJUNDAR A, HARTLEY L, MANZUR AY, KING RHM, ORRELL RW, ET. AL. A case of severe congenital chronic inflammatoy demyelinating poly-neuropathy with complete spontaneus remission. Neuromuscular Disorders. 2004;14:818-21. [ Links ]
30. VALLAT JM, TABARAUD F, MAGY L, MACIAN F. Chronic inflammatory demyelinating polirradicu-loneuropathy and their variants. Rev Neurol (Paris). 2002;158:27-31. [ Links ]
31. GORSON KC, ROOPER AH, ADELMAN LS, RAYNOR EM, SAPER CB. Chronic motor axonal neuropathy: Pathological evidence of inflammatory polyrradiculoneuropathy. Muscle Nerve. 1999;22:26670. [ Links ]
32. CORCIA P, BARBEREAU D, GUENNOC AM, DE TOFFOL B, BACQ Y. Improvement of CIDP associated with hepatitis C virus infection using antiviral therapy. Neurology. 2004;63:179-180. [ Links ]
33. KLESELER BC, DALAKAS MC, HARTUNG HP. Immune mechanisms in chronic inflammatory demy-elinating polirradiculoneuropathy. Neurology. 2002;59:7-12. [ Links ]
34. VANDER MECHÉ FGA, VAN DOORN PA. Guillain Barre syndrome and chronic inflammatory demyelinating polirradiculoneuropathy: Immune mechanisms and Update on current therapies. Ann Neurol. 1995;37 Suppl 1:14-31. [ Links ]
35. MAURER M, TOYKA KV, GOLD R. Celular immunity in inflammatory autoimmune neuropathies. Rev Neurol (Paris). 2002; 158(12 Pt 2):7-15. [ Links ]
36. TAGAWA Y, YUKI N, HIRATA K. Anti-SGPG antibody in CIDP: Nosological position of IgM anti MAG/SGPG antibody-associated neuropathy. Muscle Nerve. 2000;23:895-99. [ Links ]
37. YING YAN W, TAYLOR J, ADRIAS-KAUBA S, POLLARD J. Passive transfer of demyelination by serum of IgG from CIDP. Ann Neurol. 2000;47:765-75. [ Links ]
38. TOYKA KV AND GOLD R. The pathogenesis of CIDP: Rationale for treatment with immunomodula-tory agents. Neurology. 2003;60: 2-7. [ Links ]
39. WINER J, HUGHES S, COOPER J, BEN SMITH A, SAVAGE C. T cells infiltrating sensory nerve biopsies from patients with inflammatory neuropathy. J. Neurology. 2002;249:616-21. [ Links ]
40. SASAKI M, OHARA S, OIDE T, HAYASHIDA K, HAYASHI R. An autopsy case of chronic inflammatory demyelinating polyradiculoneuropathy with respiratory failure. Muscle Nerve. 2004;30:382-7. [ Links ]
41. FIGUEROA JP, MELLADO P. Polineuropatías en Temas de Medicina Interna. Publicación electrónica. Disponible en htp://www.escuela.med.puc.cl. [consultado el 19 de Febrero de 2004] [ Links ].
42. BENNETT JC, PLUN F, EDS. Tratado de Medicina Interna de Cecil. McGraw-Hill Interamericana Healthcare group. 1999. [ Links ]
43. SANDOVAL P, ARAYA P. Inmunología en Neurología. Cuadernos de Neurología. Vol XXIV, 2000. Disponible en http://escuela.med.puc.cl/publicacio-nes/Neurología/cuadernos/portada07.html. [ Links ]
44. MARRUECOS SANT L. Tratamiento del síndrome de Guillain-Barré mediante una dosis suficiente de gammaglobulinas. Revista electrónica de Medicina Intensiva. 2001;1(9):0 . [ Links ]
45. RADZIWILL AJ, KUNTZER T, STECK AJ. Immunopathology and treatments of Guillain Barre syndrome and chronic inflammatory demyelinating polirradiculoneuropathy. Rev Neurol (Paris). 2002;158:301-10. [ Links ]
46. CABRERA GÓMEZ JA, LÓPEZ SAURA P. Recent advances in the treatment of the nervous system disorders with interferon alfa. Rev Neurol. 1999;29:122535. [ Links ]
47. HUGHES RA. Systematic reviews of treatment for CIDP. Rev Neurol (Paris). 2002;158(12 Pt2):32-6. [ Links ]
48. GORSON KC. High dose immunosuppresion for refractory CIDP? J. Neurology. 2002;22:25-8. [ Links ]
49. BRANNAGAN TH. High dose cyclophosphamide without stem-cell rescue for refractory CIDP. Neurology. 2002;58:1856-8. [ Links ]
50. COCITO D, CIARAMITARO P, ISOARDO G, BARBERO P, MIGLIARETTI G, ET. AL. Intravenous immunoglobulin as first treatment in diabetics with concomitant distal symmetric axonal polyneuropathy and CIDP. J. Neurology. 2002;249:719-22. [ Links ]
51. WALK D, LI LYJ, PARRY GJ, DAY JW. Rapid resolution of quadriplegic CIDP by combined plasma-pheresis and IVIg. Neurology. 2004;62:155-6. [ Links ]
52. ASBURY AK. New concepts of Guillain Barre syndrome. J.ChildNeurol. 2000;15:183-91. [ Links ]
53. CABRERA LIMA AV, ESTRADA R, SANTIAGO LUIS R, ALFARO I, GONZÁLEZ A, GALARARGA INZA J. Polineuropatía crónica desmielinizante inflamatoria: una contribución a la caracaterización de la enfermedad. Rev Neurol. 1999;28:772-8. [ Links ]
54. CABRERA LIMA AV, GUTIERREZ J, ESTRADA R. Perfil electrofisiológico en la polineuropatía crónica desmielinizante inflamatoria. Rev Neurol 1999;28:353-7. [ Links ]
55. HERNÁNDEZ HERNÁNDEZ BA, FERNÁNDEZ NIN AE. Caracterización electrofisiológica de un grupo de pacientes con CIDP. Rev Cubana Invest Biomed. 2001;20:260-5. [ Links ]
56. HERNÁNDEZ HERNÁNDEZ BA, HERNÁNDEZ TORANZO R, CHARROÓ RUÍZ LE. Alteration of electrophysiological studies in a group of patients with chronic inflammatory demyelinat-ing polyneuropathy. Clin. Neurophysiol. 2001;112 (Suppl)1:40. [ Links ]
57. HERNÁNDEZ HERNÁNDEZ BA, GONZÁLEZ PILOTO MA. Electrophysiologycal characterization of a group of patients with demyelinating polineu-ropathies. Preliminary aspects. Clin. Neurophysiol. 2002;113(Suppl)1: 84. [ Links ]
58. BROMBERG MB, ALBERS JW. Pattern of sensory nerve conduction abnormalities in demyelinating and axonal peripheral disorders. Muscle and Nerve. 1993;16:262-6. [ Links ]
59. HERNÁNDEZ HERNÁNDEZ A. Contribución de la electrofisiología al diagnóstico diferencial entre el síndrome de Guillain Barré y la polineurorradicu-lopatía desmielinizante inflamatoria crónica. Rev Cubana Invest Biomed. 2008;27(3-4):0. [ Links ]
