










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Neurológica Colombiana
Print version ISSN 0120-8748
Acta Neurol Colomb. vol.29 no.4 Bogotá Oct./Dec. 2013
Esclerosis Lupoide
Lupoid sclerosis
Fernando González Trujillo, Yimy F Medina, Alfonso Lozano
Fernando González Trujillo, Neurología Clínica- Profesional observador en Neurología Oncología. Instituto Nacional de Cancerología, Hospital Simón Bolívar, Médicos Asociados Clínica Fundadores. Bogotá D.C., Colombia.
Yimy F Medina, Reumatología-Medicina Interna, Bogotá D.C., Colombia. Fundación Instituto de Reumatología.
Alfonso Lozano, Radiología-Oncología. Instituto Nacional de Cancerología, Profesor Universidad Nacional de Colombia.
Correspondencia: fernando.gonzaleztrujillo@gmail.com
Recibido: 05/08/13. Revisado: 05/09/13. Aceptado: 09/09/13.
RESUMEN
La esclerosis lupoide es una entidad de exclusión que reúne características fisiopatológicas, clínicas e imagenológicas comunes entre e lupus eritematoso sistémico (LES) y esclerosis múltiple (EM). El propósito del artículo es revisar aspectos inmunológicos, de laboratorio y diagnósticos diferenciales que faciliten comprobar la EL como una entidad clínica y definir las mejores posibilidades terapéuticas.
PALABRAS CLAVES. Autoanticuerpos, Tejido Conectivo, Lupus Eritematoso Sistémico (LES), Síndrome anti fosfolípido, Sistema nervioso Central (DeCS).
SUMMARY
Lupoid sclerosis is a diagnosis of exclusion with similar clinical, inmunological and imagenological findings between erithematous systemic lupus (SLE) and multiple sclerosis (MS). The aim of this article is to review immunology, laboratory differential diagnosis and treatment to facilitate the understanding of EL as a clinical entity.
KEY WORDS: Autoantibodies, Connective Tissue, Systemic Lupus Erythematous (SLE), Antiphospholipid syndrome (MeSH).
INTRODUCCIÓN
La Esclerosis Lupoide (EL) como entidad clínica incluye criterios diagnósticos de enfermedades como el Lupus Eritematoso Sistémico (LES) y la Esclerosis Múltiple (EM), la definición se atribuye a los Dres. Fulford y Kremer. (1) La EL surgió como propuesta para incluir los casos clínicos de LES - Síndrome Antifosfolípido y criterios de EM o con EM y anticuerpos no específicos más criterios de (similar al lupus) "lupus like" que se manifestaban como mielitis transversa y neuromielitis óptica (1).
Las manifestaciones neurológicas en la EL son diversas. Los casos sospechosos se expresan en forma completa o incompleta, fuertemente relacionados con la presencia de autoanticuerpos antifosfolípidos e incluye: ataques isquémicos focales, síntomas oculares, síndrome de Guillain Barré, convulsiones y migraña. Los casos tipo EM pueden expresar otros autoanticuerpos como los antinucleares, lo que hace algo más difícil precisar el diagnóstico diferencial (1).
La EM, el LES y el Síndrome Antifosfolípido (SAF) afectan principalmente a mujeres en edad reproductiva. Las poblaciones negra y japonesa se afectan poco por la EM pero tienen incidencias altas de LES (2). El LES afecta las mujeres según norma de ANC entre 20 y 60 años de edad con una incidencia de 1 en 700, y la razón mujer hombre es 10:1 (3).
El trasfondo inmunológico y la evolución similar clínica observada en la EM y el LES, con remisión y con recaídas, con manifestaciones neurológicas y lesiones multifocales en la sustancia blanca documentadas por resonancia magnética conlleva un desafío diagnóstico y terapéutico (2,3).
El propósito de este escrito es revisar aspectos inmunológicos, de laboratorio y de diagnóstico diferencial que faciliten identificar la EL como una entidad clínica y definir las mejores posibilidades terapéuticas según el caso clínico.
Patogénesis
La EM y el LES, son enfermedades autoinmunes en las que células T CD4 del subtipo TH1 y TH17 reaccionan contra antígenos propios de la mielina produciendo la activación de macrófagos y la inflamación característica en el sistema nervioso central. Aún es un enigma cómo se activan las células, y se plantean causas infecciosas virales que activan las células T contra la propia mielina mediante el fenómeno de "imitación molecular" o mimetismo molecular. Se han informado unas funciones defectuosas de las células T reguladoras (3). Cuando las células T específicas contra la mielina se activan migran hacia el sistema nervioso central y una vez en contacto con las proteínas de la mielina liberan citocinas que activan macrófagos y otras células T generando lesión. Hay informes de polimorfismo genético que incluye el locus HLA-DR2 como relación más fuerte (3-5).
El compromiso en la sustancia gris del SNC es común en muchas enfermedades inflamatorias y la EM figura entre ellas. Los trabajos de Lucchinetti y colaboradores en el año 2011 revelaron que en fases tempranas de la EM hay lesión en la sustancia gris que se incrementa con la duración de la enfermedad (4). Los mecanismos causales para la inflamación y la degeneración neuronal de la sustancia blanca son diferentes: se propone una inflamación meníngea subpial focal como evento crucial para la demielinizacion cortical por un ataque mediado por las células T que altera la barrera hematoencefálica y así permite el paso de anticuerpos. Además, el sistema inmune humoral media el proceso inflamatorio persistente. La reacción patogénica activa los astrocitos, la microglia y los oligodendrocitos con persistencia de las células inmunes en las meninges lo que finalmente conduce a la reacción inflamatoria crónica.
El lupus es una entidad con alteración de la tolerancia a los propios tejidos por parte de los linfocitos T y B y está determinada por factores genéticos y ambientales. Entre los factores genéticos se encontró que la herencia de alelos HLA del tipo DR2 y DR3 aporta una probabilidad mayor de desarrollar la enfermedad cuando ambos se presentan en un mismo individuo (3). Otro factor descrito es la deficiencia en las proteínas del complemento de la vía clásica tipo C1q, C2 o C4 (3). Como causas ambientales se propone la exposición a la luz ultravioleta que incrementa la apoptosis celular y la liberación secundaria de antígenos nucleares (3,4).
Otras hipótesis que explican el LES, describen además, al interferón alfa de las células dendríticas plasmocitoides que se producen anormalmente en grandes cantidades y la participación de receptores similares al tipo Toll (like TLR) que reconocen al DNA y al RNA; especialmente el TLR9 muy importante en la activación de células B específicas contra los antígenos nucleares propios (3). En el lupus neuropsiquiátrico hay hipótesis que asignan un rol patogénico a los autoanticuerpos contra DNA de doble cadena (dsDNA) y las subunidades NR2A y NR2B de los receptores ionotrópicos que se activan por glutamato (NMDA-R). Con el compromiso cognoscitivo difuso en los pacientes se ha reportado un mecanismo patogénico alternante de los anticuerpos antifosfolípidos, la demostración "in vitro" de la producción intratecal de los anticuerpos puede evidenciar una posible modulación de la función neuronal (3-7).
Con respecto al síndrome antifosfolípido, los anticuerpos ocasionan una reacción cruzada con la mielina o las proteínas relacionadas y los fosfolípidos (cefalina, esfingomielina) del cerebro y se informa de un compromiso neurológico semejante a la EM por procesos de "semejanza o mimetismo molecular" con la mielina y otros antígenos que producen eventos trombóticos microvasculares, vasculopatía o vasculitis autoinmune similar al LES (2,3).
DIAGNÓSTICO
Realizar el diagnóstico diferencial entre la EM y el LES- SAF no es fácil incluso con las manifestaciones clínicas, con hallazgos de imágenes y de laboratorio. La evolución clínica que cursa con períodos de recaída-remisión es indistinguible entre las dos enfermedades. Para el proceso diagnóstico no se tienen pruebas patognomónicas que definan plenamente cada entidad (2,3,6) (Tablas 1 y 2).
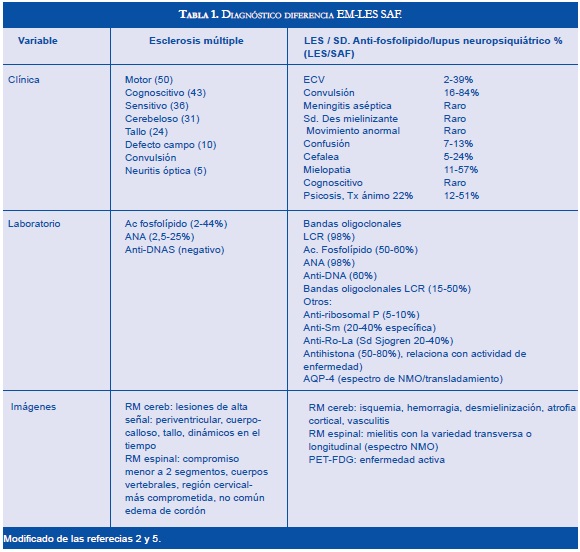

La EM se diagnostica con la evidencia objetiva de compromiso del sistema nervioso central diseminado en espacio y tiempo, con varios episodios y más de un área afectada. Los síndromes neurológicos aislados son un reto diagnóstico ya que en el 90% de casos la EM se manifiesta aisladamente y en el LES-SAF pueden ser la única manifestación clínica (1,2).
El LES asociado al SAF y la EM indistintamente pueden manifestarse con mielitis transversa, neuritis óptica aislada o asociada con mielitis transversa, síndromes cerebelosos o de tallo cerebral, diplopía o neuralgia del trigémino. Las causas propuestas para explicar las manifestaciones neuropsiquiátricas en el LES son mecanismos demielinizantes comparables a los de la EM, y la atrofia cerebral es similar en las imágenes de resonancia magnética cerebral seriada (6,7). El retroceso cognoscitivo se asocia a la EM y los tratamientos inmunomoduladores iniciados tempranamente reducen su avance (6). Con los anticuerpos antifosfolípidos se mencionan alteraciones en la función cognoscitiva en adición a los efectos asociados con las lesiones vasculares y las convulsiones (6- 8).
La Neuromielitis Óptica (NMO) se ha relacionado con la EM y el LES, en la actualidad se considera una entidad separada de la EM con patogénesis diferente que predomina en el sexo femenino. Se encuentra una reactividad mediada por la inmunoglobulina del tipo G contra su principal antígeno identificado la acuaporina-4 (AQP-4). La NMO se puede manifestar en forma agresiva en el LES (1-3). Jarius y colaboradores informaron frecuencias altas de anticuerpos AQP-4 en pacientes con enfermedades del tejido conectivo y cuando se evaluaron los pacientes con LES o síndrome de Sjögren con clínica de neuritis óptica o mielitis los títulos de AQP-4 fueron altos. El mecanismo determinante de la asociación con LES o Síndrome de Sjögren en la EM sigue sin resolverse pero con los datos actuales se recomienda valorar la presencia de anticuerpos AQP-4 en los pacientes que llenen criterios para la NMO (8,9). La EM puede tener anticuerpos antifosfolípidos positivos y manifestarse con NMO y la neuritis óptica asociada con LES y SAF suele ser unilateral al contrario de la EM en la cual puede ser bilateral (2,3).
El LES con mielitis transversa tiene una mayor prevalencia de anticuerpos antifosfolípidos que en los casos de LES sin mielitis transversa. La "mielitis longitudinal" principalmente cervical y torácica que describe el compromiso continuo multinivel se presenta principalmente en pacientes con LES (1,2). La mielitis en la EM afecta usualmente un nivel o de manera alterna varios niveles.
En el LES hay compromiso del sistema nervioso periférico lo que no sucede con la EM. Otras manifestaciones que pueden ayudar al enfoque hacia un LES/SAF son eventos trombóticos, abortos, morbilidad en embarazo como síndrome HELLP, "livedo reticularis" y trombocitopenia. La foto sensibilidad, la erupción dérmica, las artralgias y el síndrome seco dirigen hacia enfermedades reumáticas. En el Síndrome de Sjögren el compromiso del sistema nervioso central es poco frecuente pero se han descrito casos de neuritis óptica, mielitis longitudinal y lesiones en la sustancia blanca similares a la EM. Las denominadas enfermedades del tejido conectivo o reumáticas sobrepuestas (es decir características de 2 o más enfermedades) tienen como principal manifestación síntomas neurológicos, miositis y otros síntomas similares a los presentados con LES, Síndrome de Sjögren, que tienen una frecuencia de presentación y mejor pronóstico (1-3,8).
No se cuenta con pruebas específicas para diferenciar entre las dos enfermedades, los métodos más útiles son la resonancia magnética (RM) de cerebro y medula espinal, la valoración del líquido cefalorraquídeo que incluye el índice de inmunoglobulinas (IgG) con inmunoelectroforesis, los potenciales evocados visuales y el perfil de autoanticuerpos séricos.
La RM (Figuras 1-4) permite valorar mejor la sustancia blanca cerebral y se pueden ver lesiones pequeñas de causas isquémicas que se presentan en el LES/SAF hasta en 30 a 70% y se asemejan a las placas desmielinizantes de la EM (2). El concepto de realce con gadolinio que sugiere inflamación no es específico para la EM, los complejos inmunes inducen la alteración de la barrera hematoencefálica, de modo que la RM como método diagnóstico no permite definir la causa. La distribución de las lesiones pueden orientar al diagnóstico y las lesiones subcorticales prevalecen en el LES/SAF y las periventriculares próximas al cuerpo calloso como lesiones ovoides (dedos de Dawson) y los hoyos negros son característicos de la EM. La evolución de las lesiones en diferentes secuencias de RM es de ayuda para la diferenciación y se consideran de evolución dinámica en la EM y en cambio permanecen estáticas en el LES con o sin SAF y mejoran con la anticoagulación (2).
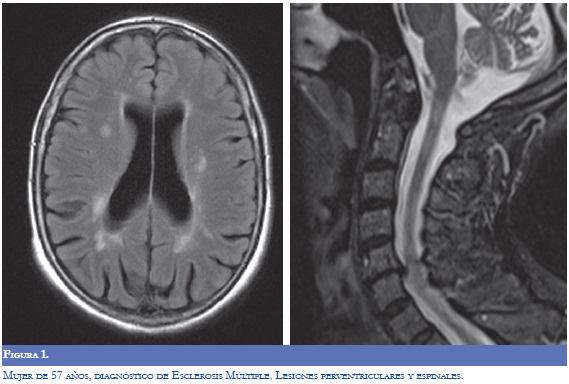
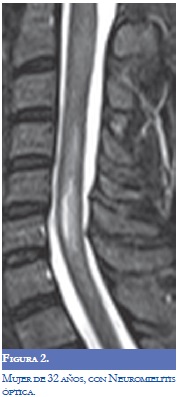
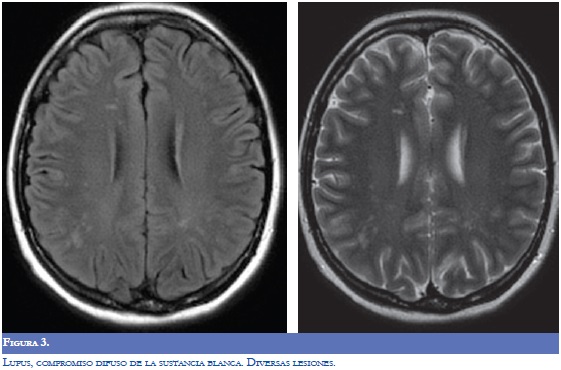
En relación a la atrofia cerebral reportada en estas entidades por la RM, en el LES se mostró una reducción en el volumen cerebral y del cuerpo calloso que se asoció con la duración de la enfermedad, con el compromiso cognoscitivo y con otras manifestaciones del sistema nervioso central; el uso y la dosis de esteroides se asoció más con atrofia del hipocampo (7,8).
La espectroscopia por RM aplicada en el LES en las fases agudas y quiescentes revela anormalidades metabólicas de las sustancias gris y blanca que por RM convencional se informan como normales. Se consideran que es por daño o pérdida neuronal o por desmielinizacion. La tomografía por emisión de positrones con fluorodeoxiglucosa (PET-FDG) en el LES activo o quiescente ha revelado anormalidades tanto en la sustancia gris como en la blanca, en áreas cerebrales prefrontales, en la parietal superior e inferior, la parietoccipital, la temporal superior y la occipital. La PET-FDG mostró en el LES activo un metabolismo disminuido aproximadamente en el 60 a 80% de los casos de las áreas parietoccipitales bilaterales en contraste con los estudios por RM convencional que fueron normales (6,8).
Los estudios de líquido cefalorraquídeo (LCR) permiten valorar directamente la causa inflamatoria tanto en la EM como en el LES hay leve pleocitosis linfocítica, un índice alto de IgG y bandas oligoclonales positivas. La ausencia de bandas oligoclonales en LCR hace poco probable el diagnóstico de EM (2,6). Los potenciales evocados visuales se consideran como una herramienta útil de diagnóstico y un resultado normal es inusual en EM (2,8).
Los anticuerpos antinucleares (ANAS) en estas 2 entidades son inespecíficos y se encuentran también en personas sanas en 2 a 8% y en muchas otras enfermedades. Los valores altos y persistentes pueden ayudar a pensar en una enfermedad del tejido conectivo o reumático.
Los anticuerpos antifosfolípidos se asocian con alto riesgo de compromiso cardiovascular de tipo trombótico y la interpretación de los resultados debe ser cuidadosa, pues, en la población general se informan títulos positivos en 2% a 5% sin que haya síntomas. Otras condiciones médicas que dan positividad en estos anticuerpos son: las infecciones, las neoplasias, algunos medicamentos, las enfermedades autoinmunes como el LES principalmente (36%) con compromiso del SNC (50% al 60%) la Miastenia Gravis, el Síndrome de Eaton-Lambert, la migraña, la EM y la Neuromielitis Óptica. Los títulos de los anticuerpos son fluctuantes y en las fases agudas pueden ser negativos (2).
Diagnóstico diferencial
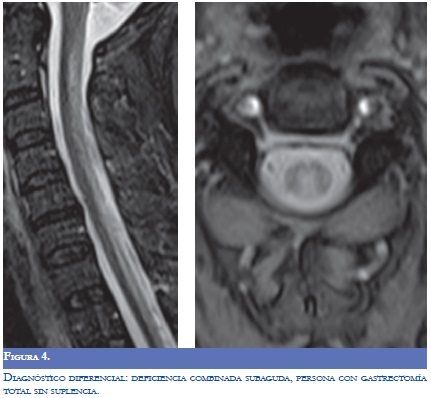
Una consideración importante en la aproximación diagnóstica consiste en descartar que el síndrome clínico sospechado no se relacione con disfunción orgánica múltiple asociada con patologías agudas, por procesos infecciosos, con efectos secundarios de medicamentos, anormalidades metabólicas como la uremia y otras (Figura 4) (8). Otras entidades para tener presente son: la leucoencefalopatía multifocal progresiva, la tuberculosis, la sífilis, la demielinizacion por virus como el VIH, el virus de sarampión asociado con la Pan Encefalitis Esclerosante Subaguda (PEESA) (4). La infección óptica. por el virus Linfotrópico de célula T humana tipo l (HTLV I) se asocia con una mielopatia inflamatoria crónica que clínicamente se manifiesta como una mielitis transversa. La infiltración linfocítica perivascular es el marcador histológico, se infectan los linfocitos CD4 que pueden detectarse en el sistema nervioso. La evolución crónica y la progresión lenta dificultan el diagnóstico diferencial con la EM y otras causas de mielopatía; se reporta que la medición simultánea de la carga viral de HTLVI en el LCR y en células mononucleares de sangre periférica ayuda a depurar el diagnóstico, el incremento en la relación con manifestaciones clínicas agudas orienta hacia esta entidad viral (10).
Las talasemias producen eventos isquémicos que cursan con patrones de remisión-recurrencia y cambios en las imágenes de resonancia magnética que también son característicos de la EM (11).
El síndrome de Susac's se asocia con vasculitis, complicaciones neurológicas que afectan preferentemente el cerebro, la retina y el oído interno relacionados con microangiopatías y oclusiones de ramas arteriolares y se proponen alteraciones en la coagulación, microembolismos o ambos (11).
La coartación aórtica produce compromiso neurológico por infartos cerebrales resultado de eventos embólicos desde la aorta dilatada proximal a la obstrucción, además de cefalea, episodios de inconciencia, hemorragia intracerebral, compresión e isquemia de cordón espinal.
La CADASIL (Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy) se produce por la mutación o deleción en el gen Notch3 del cromosoma 19q12 y patológicamente se caracteriza por el hallazgo de un material granular osmiofílico que se deposita en las paredes vasculares. Las manifestaciones neurológicas pueden ser focales o múltiples y evolucionan con recaídas o en forma progresiva y los hallazgos por RM pueden simular los reportados en la EM (11).
TRATAMIENTO
Los planes terapéuticos se enfocan según la certeza diagnóstica que se posea. Los tratamientos de la EM con interferones pueden potencialmente inducir o empeorar un cuadro clínico de actividad lúpica cuando no se diagnóstica adecuadamente (12).
En LES el plan de tratamiento es un reto debido a la dificultad para precisar el mecanismo determinante en cada caso clínico y se puede considerar la terapia dual de inmunosupresión y de anticoagulación. Los esteroides se usan en las fases agudas tanto del LES como de la EM.
La anticoagulación para pacientes con demielinizacion y persistencia de anticuerpos antifosfolípidos es un interrogante que no tiene respuesta precisa, si presenta SAF se escoge una óptima anticoagulación con warfarina (llegando a un INR de 3 a 4), en tal caso el uso de profilaxis inicial con antiagregación plaquetaria o las dosis subóptimas de anticoagulación oral son inapropiadas (1). Se han reportado pacientes con diagnóstico y clínica parecida a la esclerosis múltiple de EM tipo "like" con o sin LES asociado con significativa mejoría clínica y radiológica después de la anticoagulación (1,2).
Para casos clínicos complejos refractarios a los tratamientos referidos, se ha descrito el uso de la plasmaferesis, la combinación de inmunoabsorción y pulsos endovenosos de esteroides de rituximab o el transplante autólogo de células madre (1- 3,8,9).
La EL no está definida como una entidad clínica en la actualidad, el término propuesto agrupa casos clínicos que no cumplen con los criterios de diagnóstico para LES/SAF y EM. Son entidades complejas que comparten una causa inmune, evolución y planes de tratamiento similares pero que son patologías diferentes; en el espectro de las enfermedades desmielinizantes la Neuromielitis Óptica (NMO) se clasifica aparte de la EM en los años recientes (9). Los "síndromes clínicos aislados inmunes" que se manifiestan en las dos entidades conforman un reto diagnóstico y terapéutico (2,9).
Surgen interrogantes con los pacientes que tienen anticuerpos antifosfolípidos positivos y manifestaciones clínicas similares al LES o a la EM tipo neuritis óptica, mielitis transversa o longitudinal, respecto de si deben diagnosticarse como EL o como un síndrome de traslapamiento de las entidades. Deben realizarse análisis secuenciales para depurar los diagnósticos; con anticuerpos antifosfolidos persistentes positivos la terapia anticoagulante evidenció en los pacientes una buena respuesta, lo que es un fuerte argumento para responder la pregunta planteada (7,13). Debe esclarecerse mejor la participación de los autoanticuerpos en la patogénesis de estas entidades o establecer si sólo son un epifenómeno clínico.
Los métodos de diagnóstico por imágenes funcionales deben ser complementarios a los estudios actuales de resonancia, repetirse en el tiempo para documentar mejor los procesos dinámicos que se suceden y así aclarar interrogantes.
El diagnóstico diferencial de la EM con manifestaciones "atípicas" debe realizarse siempre con LES/SAF, sin olvidar el espectro de las entidades clínicas con manifestaciones similares y para aquellos casos clínicos complejos es el proceso secuencial de seguimiento lo que mejora los diagnósticos.
REFERENCIAS
1. KEISERMAN B, GARCIAS LF, DA SILVA LF, KEISERMAN MW, VON MÜHLEN CA, STAUB HL. Lupoid sclerosis. Rheumatol Int. 2010; 30:431-4. [ Links ]
2. FERREIRA S, D'CRUZ DP, HUGHES GRV. Review Multiple sclerosis, neuropsychiatric lupus and antiphospholipid syndrome: where do we stand?. Rheumatology. 2005; 44:434-42. [ Links ]
3. ABBAS AK, LICHTMAN AH, PILLAI S. Cellular and Molecular Immunology, Philadelphia: Elsevier Saunders. 2012:407-23. [ Links ]
4. JUNKER A, BRU¨CK W. Auto inflammatory grey matter lesions in humans: cortical encephalitis, clinical disorders, experimental models. Curr Opin Neurol. 2012; 25:349-57. [ Links ]
5. GOLD R, MONTALBAN X. Multiple sclerosis: more pieces of the immunological puzzle. The lancet neurology. 2012; 11:9-10. [ Links ]
6. VOSS EV, STANGEL M. Nervous system involvement of connective tissue disease: mechanisms and diagnostic approach. Curr Opin Neurol. 2012; 27:306-15. [ Links ]
7. SOUIRTI Z, LAHLOU M, OUALI OE, CHTAOU N, AARAB C, ET AL. Neuropsychiatric Systemic Lupus Erythematous. Open J Rheumatol Autoim Diseass. 2013; 3:86-91. [ Links ]
8. MUSCAL E, BREY RL. Neurologic Manifestations of Systemic Lupus Erythematous in Children and Adults. Neurol Clin. 2010; 28:61-73. [ Links ]
9. WINGERCHUK DM, LENNON VA, LUCCHINETTI CF, PITTOCK SJ, WEINSHENKER BG. The spectrum of neuromyelitis optica. The Lancet Neurology. 2007; 6:805-15. [ Links ]
10. HAYASHI D, KUBOTA R, TAKENOUCHI N, NAKAMURA T, FUJIO UMEHARA F, ARIMURA K, ET AL. Case Report Accumulation of human T-lymphotropic virus type I (HTLV-I), infected cells in the cerebrospinal fluid during the exacerbation of HTLV-I_associated myelopathy. J NeuroVirology. 2008; 14:459-63. [ Links ]
11. KAIMEN-MACIEL DR, CALLEGARO D. Unusual Clinical Cases that Mimic MS: A report on selected cases presented at the MS Forum /Interactive Symposium at the LACTRIMS Congress, 27 August 2004, Iguazu Falls, Brazil. The International MS Journal. 2006; 13: 77-83. [ Links ]
12. MEDINA Y, MARTÍNEZ J, FERNÁNDEZ A, QUINTANA G, RESTREPO J, RONDÓN F, IGLESIAS A. Asociación de lupus eritematoso sistémico y esclerosis múltiple: Esclerosis lupoide. Descripción de 4 casos. Revista Colombiana de Reumatología. 2010; 17:111-22. [ Links ]
13. FERREIRA S, D'CRUZ DP, HUGHES GRV. Multiple sclerosis, neuropsychiatric lupus and antiphospholipid syndrome: where do we stand?. Rheumatology. 2005; 44:434-42. [ Links ]
